This chapter should be cited as follows:
Vijayaraghavan SB, Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.420463
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 18
Ultrasound in obstetrics
Volume Editors:
Professor Caterina M (Katia) Bilardo, Amsterdam UMC, Amsterdam and University of Groningen, Groningen, The Netherlands
Dr Valentina Tsibizova, PREIS International School, Florence, Italy

Chapter
Ultrasound of the Fetal Gastrointestinal Tract
First published: January 2024
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

INTRODUCTION
Prenatal diagnosis of gastrointestinal tract (GIT) anomalies has a significant impact on the management of these conditions by allowing delivery in centers in which prompt surgical intervention is available. Furthermore, the couple can be counseled about the management of the pregnancy before delivery and of care of the newborn after delivery. There is a paucity of studies in the literature about this subject since they manifest late in pregnancy which may be due to later occurrence of insult or delayed manifestation due to delayed occurrence of effective swallowing and peristalsis or both.
The GI tract is composed of the esophagus, stomach, and duodenum, small and large bowel. Most of these structures are contained in the abdomen and hence are scanned as part of fetal abdomen. Unlike the other organ systems which have extended protocols such as fetal echocardiogram and fetal neurosonogram, there is no extended protocol described for fetal GIT system to date. Structural anomalies of the fetal GIT are not seen on prenatal ultrasound per se, but they are seen and diagnosed by secondary effects such as obstruction. Ultrasound visualization of normal fetal GI tract depends on gestational age. There has been interest in prenatal discussion of anomalies of GIT only recently, since most of them are not seen during the second trimester anomaly scan and they are manifest only in late second and third trimester. This is because effective peristalsis of the stomach and bowel occurs during this time and secondary effects due to accumulation of fluid only occurs then. It is also due to the fact that many bowel anomalies are as a result of vascular insults which happen late in gestation.
There are two aspects for fetal GIT:
- Structure – The only structure of fetal GIT which is seen on routine second trimester anomaly scan is the fetal stomach.
- Function – There are two functions of the fetal GIT:
- Propulsive action by peristalsis which takes the swallowed amniotic fluid up to the small bowel;
- Absorption – the amniotic fluid is absorbed through the fetal small bowel.
Congenital anomalies can be classified based on whether the defect is structural or functional. Structural anomalies result from either defective embryogenesis or intrauterine complications, such as ischemia. Functional defects have normal anatomy but disrupted flow of GIT contents. In most cases, structural defects adversely impact functional capability and manifest due to impairment of function.
The sonographic appearance of normal fetal GIT is dynamic. It varies with gestational age and also during the period of scan due to contents and peristalsis. This is a unique feature of fetal GIT.
TECHNIQUE OF SONOGRAPHY OF FETAL GIT
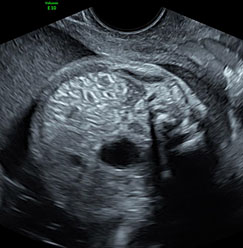
Since most part of GI tract is contained in abdomen, anomalies of GIT are seen in sections of fetal abdomen. Almost all of the guidelines for anomaly scan describe the structures to be seen in abdomen.1,2,3 They do not describe the required sections of anatomy to be obtained unlike the fetal heart or central nervous system. The same can be achieved by the three axial sections obtained as part of second trimester anomaly scan (STAS) – namely upper and mid-abdomen, pelvis and a coronal scan of the abdomen.
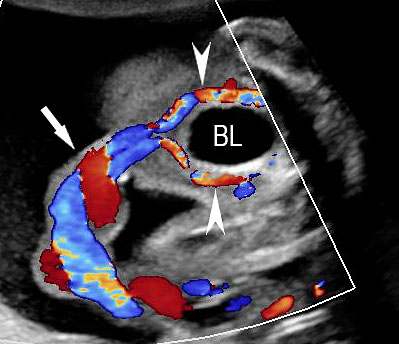
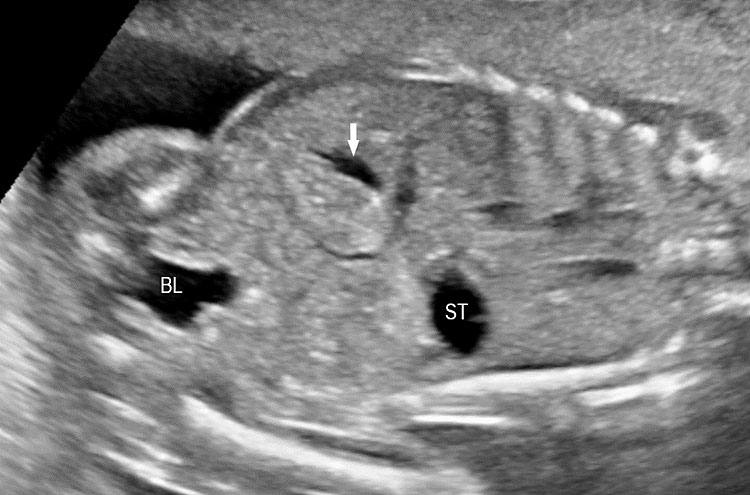
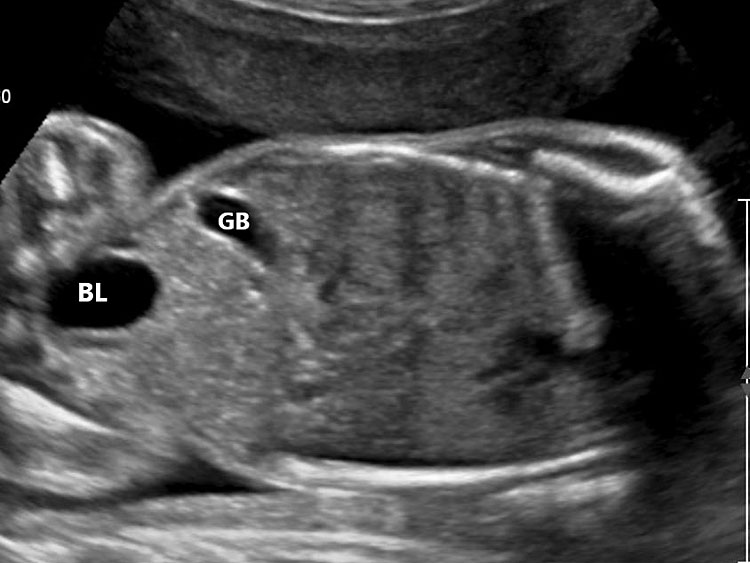
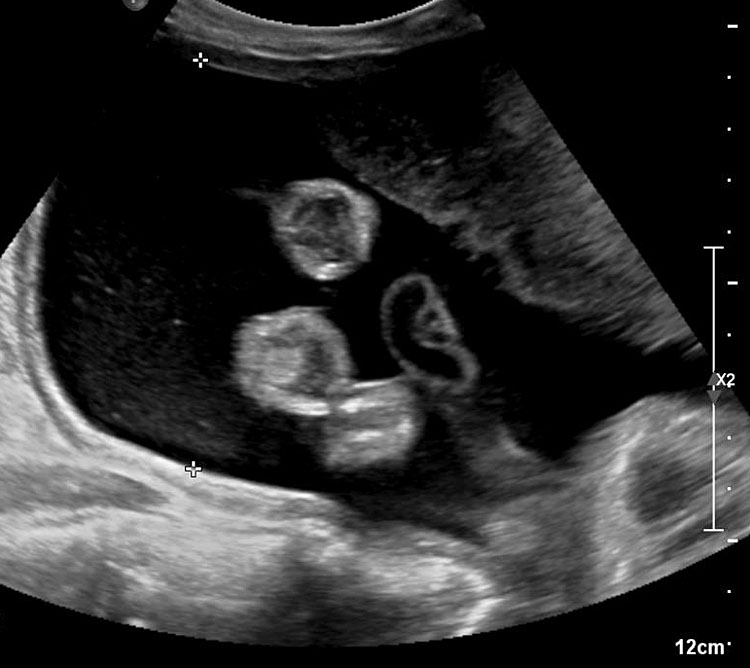
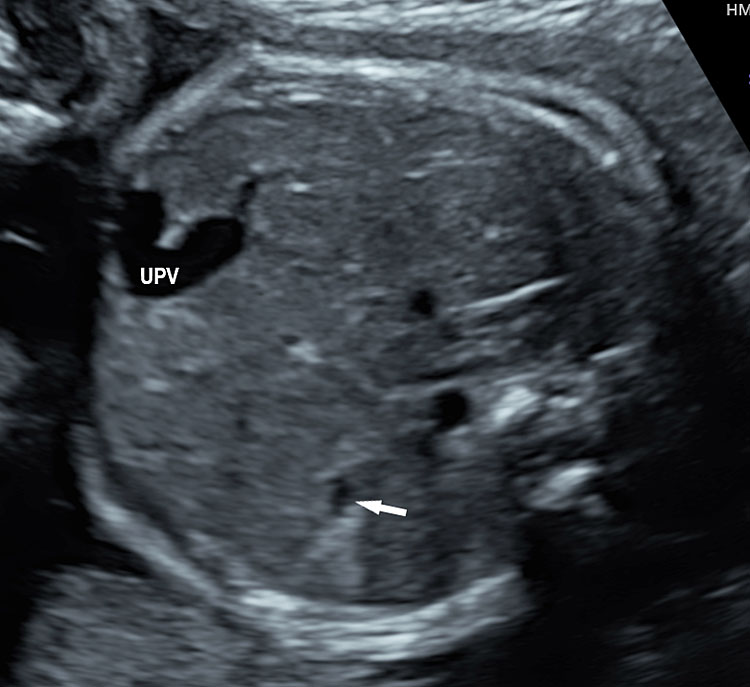
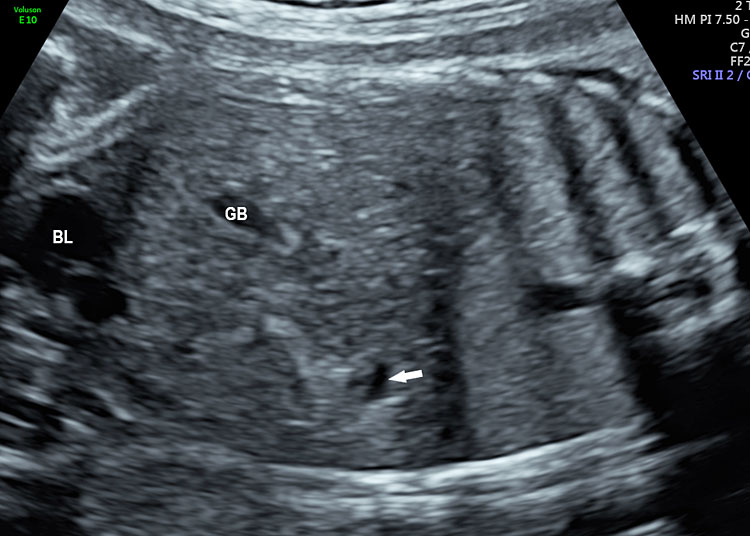
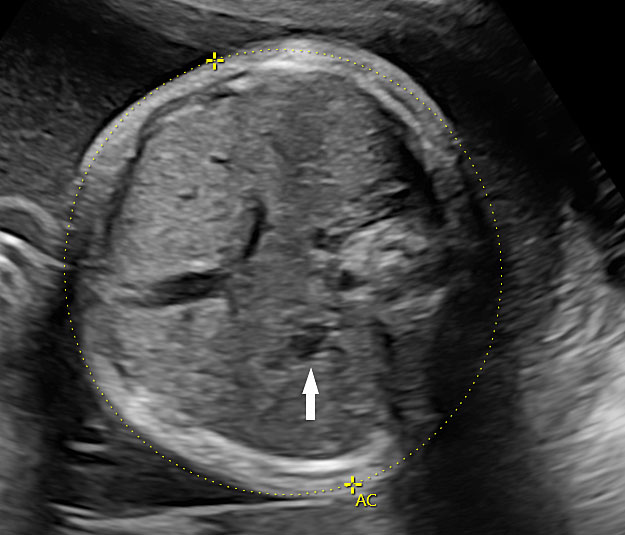
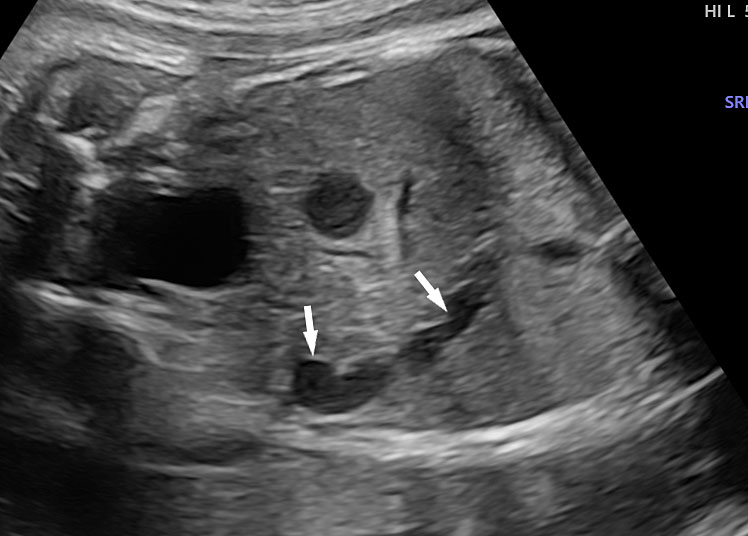
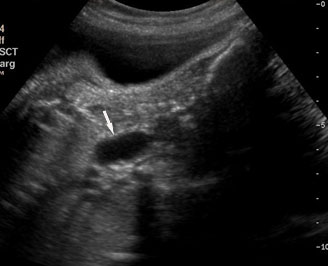
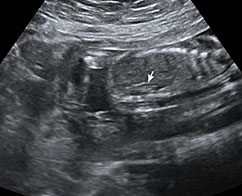
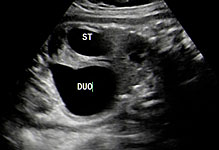
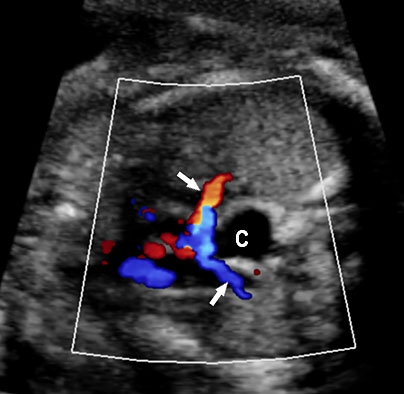
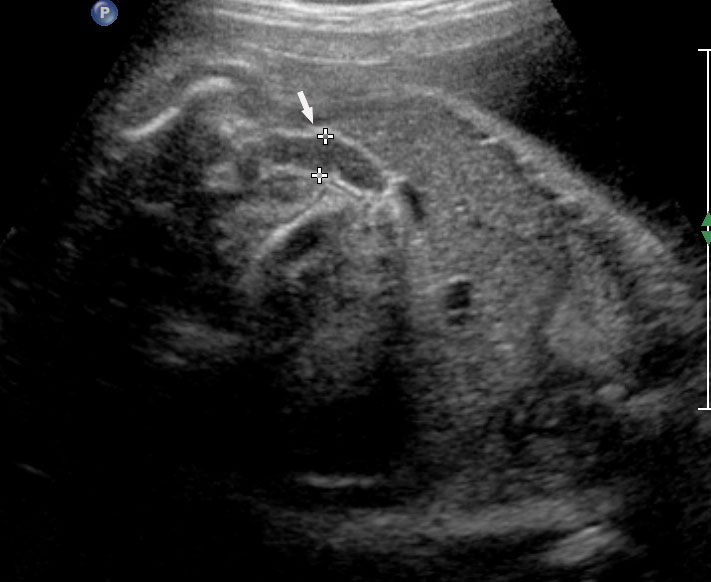
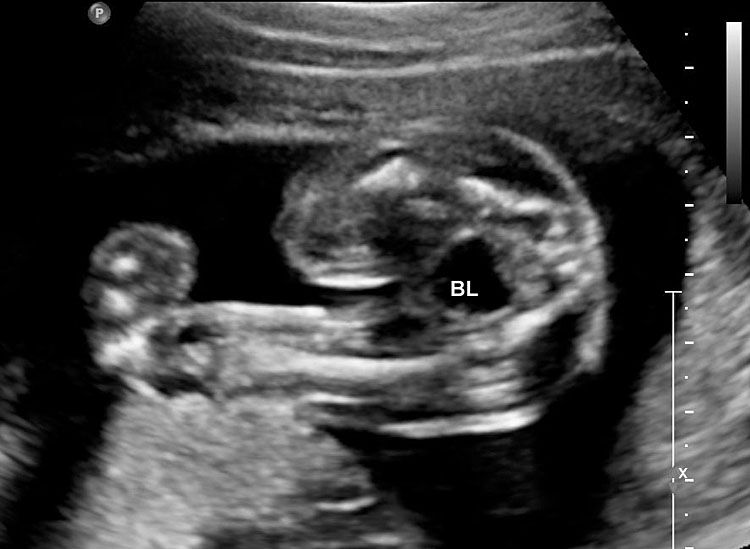
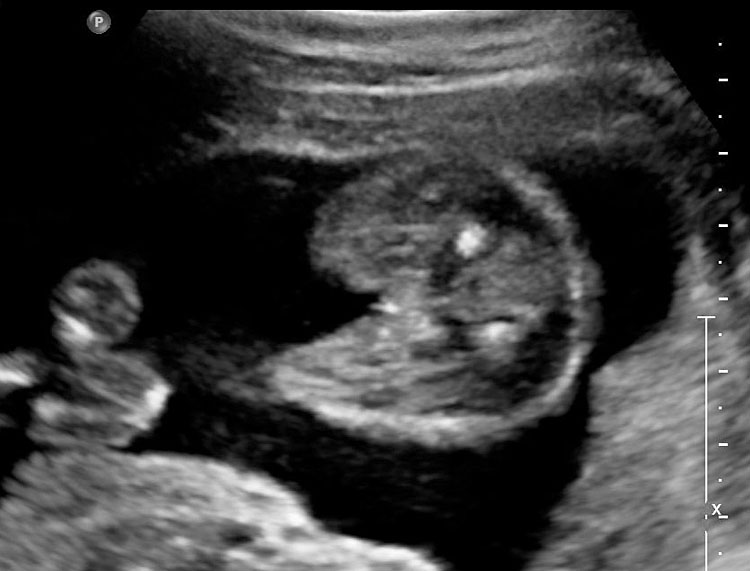
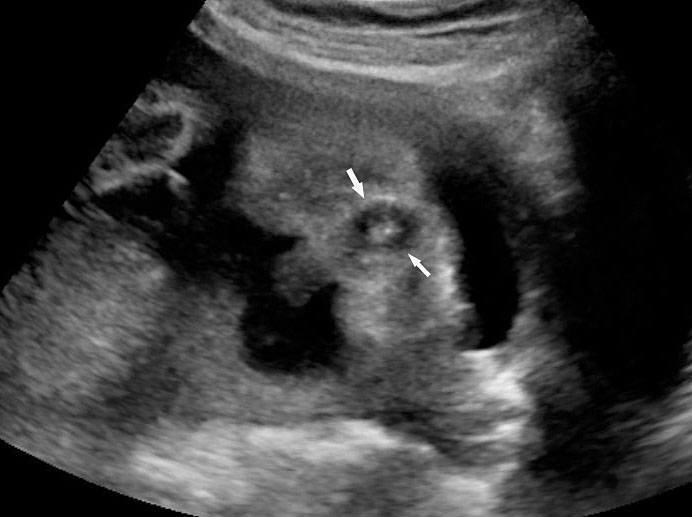
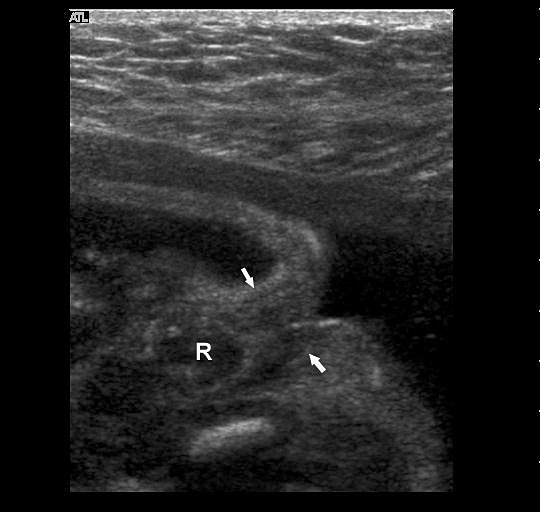
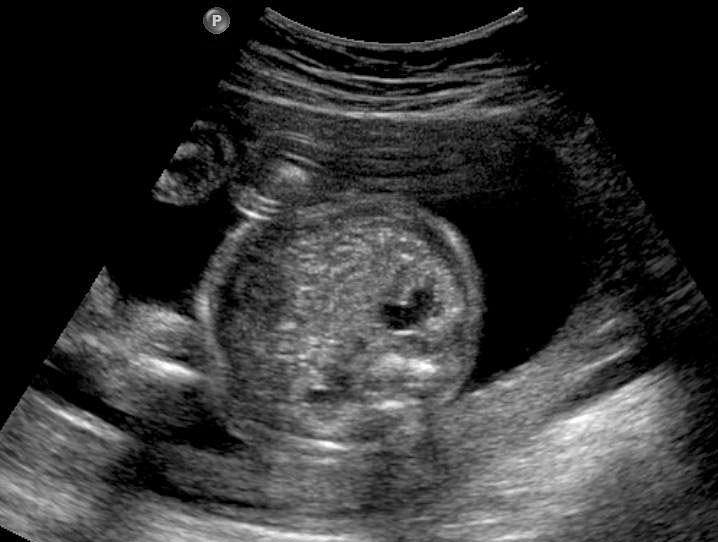
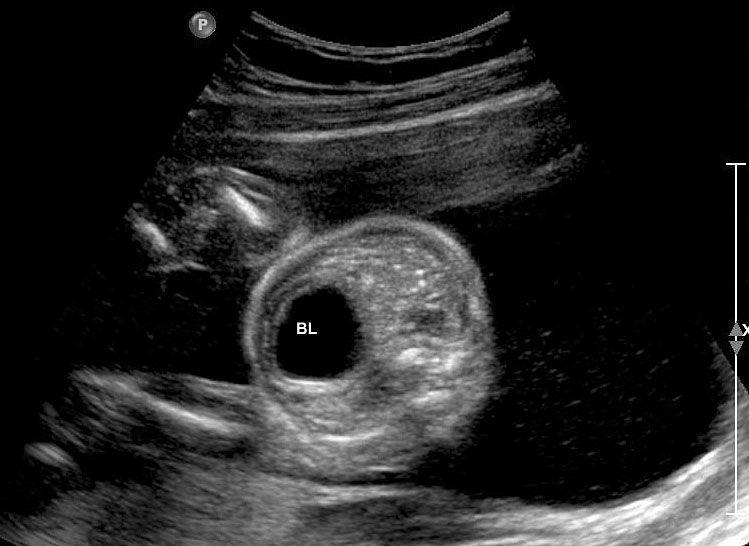
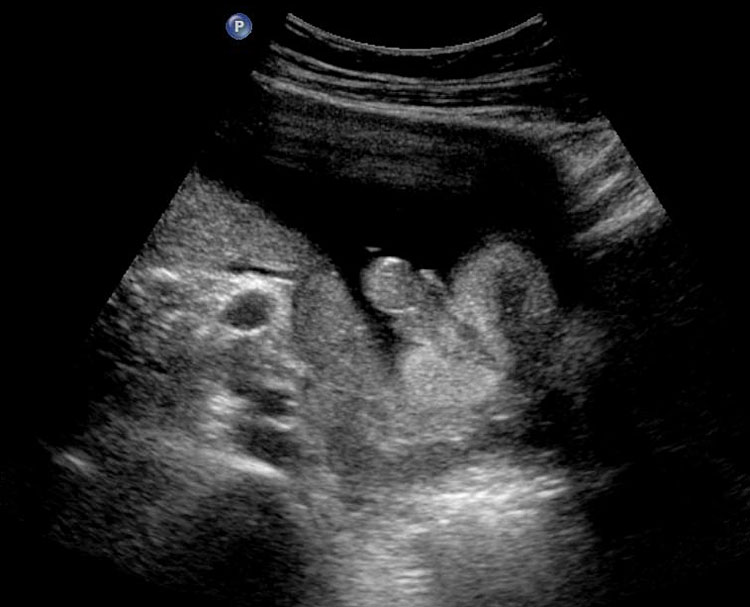
The first section is the axial section of upper abdomen along which the abdominal circumference is measured (Figure 1). In this section the structures to be studied are the spine posteriorly, the umbilico-portal vein seen as a hockey stick formed by the anteroposteriorly oriented left branch and horizontal right branch of portal vein, the stomach bubble, the liver, spleen, aorta and inferior vena cava. In this section abdominal organ situs is determined. The stomach bubble should be on the left side. The fetal stomach is seen as an oval or round echo-poor structure because of fluid distending it. The size of stomach bubble is variable due to gastric emptying and filling, owing to swallowing of amniotic fluid. If stomach bubble is not visualized, the scan should be repeated after an interval. The second section is taken to show the fetal insertion site of umbilical cord to rule out abdominal wall defects (Figure 2). This section will also show the mid-abdomen containing the small bowels which are featureless in the scan using conventional probe. The third section is axial scan of pelvis (Figure 3A). The urinary bladder is seen as an oval fluid=filled structure in this section. It shows filling and emptying cycles due to micturition. When urinary bladder is not seen, the scan has to be repeated since the urinary bladder may be empty due to fetal micturition. With color Doppler study, in a slightly oblique scan of urinary bladder including the cord insertion, the two umbilical arteries are seen to skirt around the urinary bladder (Figure 3B,C). When urinary bladder is not seen the umbilical arteries help to confirm that the section is appropriate. The last section in protocol is the coronal scan of the abdomen. It will show three fluid-filled structures – the stomach in left upper abdomen, the gall bladder in the right upper abdomen and the urinary bladder in the midline in the pelvis (Figure 4). Any other additional fluid-filled structure will need further evaluation. These are the standard sections of abdomen to be obtained to see the structures as per guidelines. When an abnormal appearance is seen, the scan has to be extended appropriately.
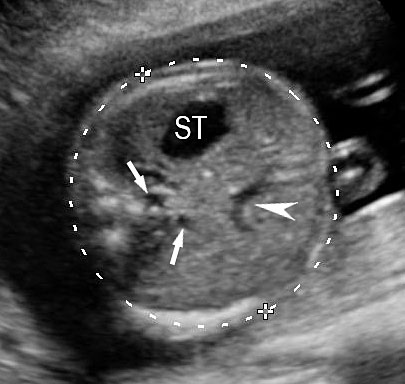
1
Axial section of fetal abdomen along which the abdominal circumference is measured. The structures seen are stomach bubble (ST) on left side, umbilico portal vein (arrowhead), aorta on left side and inferior vena cava on right side (thin arrows).
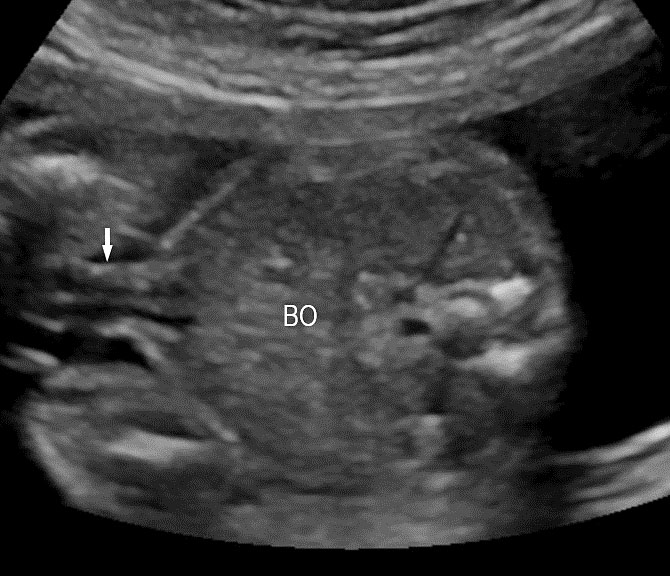
2
Axial section of mid-abdomen showing the fetal cord insertion (arrow) and small bowels (BO) filling the abdomen.
(A) |
(B) |
(C) | |
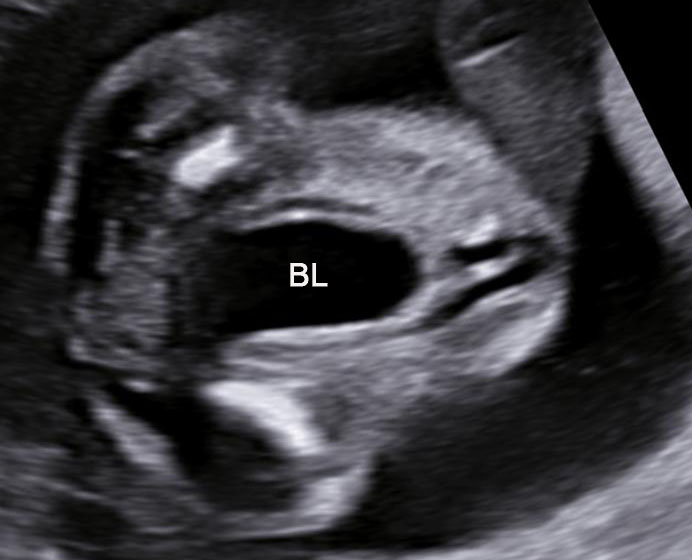
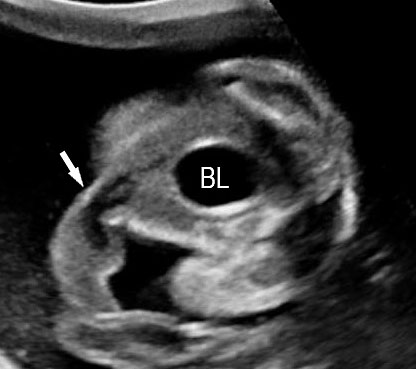
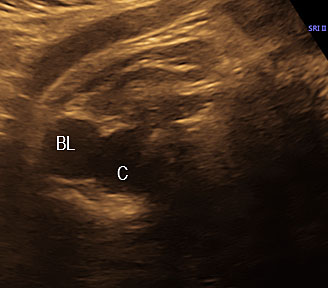
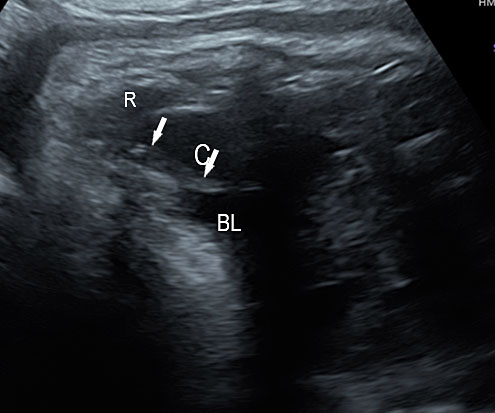
3
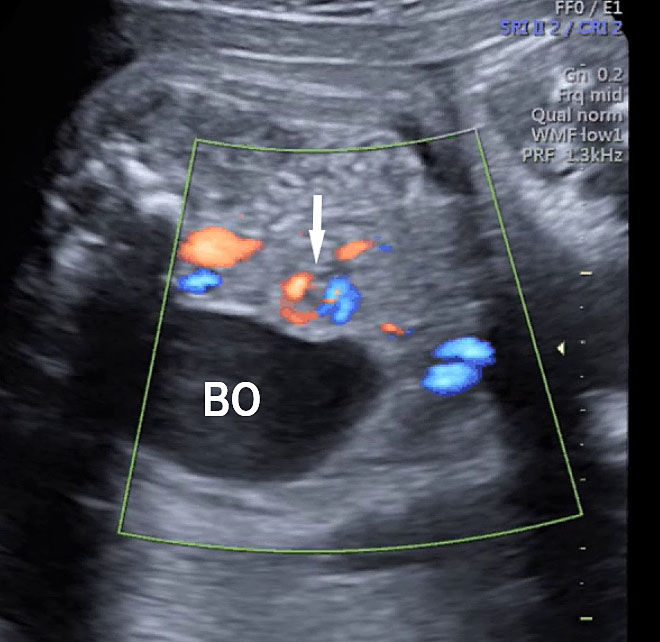
(A) Axial section of fetal pelvis showing the fluid-filled urinary bladder (BL). (B) Gray scale. (C) Color Doppler oblique section of pelvis and cord insertion (arrow) showing the urinary bladder (BL) and the two umbilical arteries (arrowheads) skirting on either side of bladder.
4
Coronal scan of fetal abdomen showing the three fluid-filled structures – stomach (ST), gall bladder (arrow) and urinary bladder (BL).
The resolution of the ultrasound scanners have improved remarkably over the years. Hence the visualization of abdominal structures has improved to a great extent. The highest frequency probe, allowing adequate penetration for the structure to be seen, has to be used. The high-frequency linear transducer has to be used to see better details of organs whenever possible. This may need a repeat scan to allow the fetus to move, so that the organ comes nearer the anterior abdominal wall. The improved resolution of this technique and its utility is illustrated in the subsequent sections.
Fetal GIT and amniotic fluid dynamics
The amniotic fluid is vital for the well-being of the fetus. There is an association between the amniotic fluid dynamics and fetal GIT. Membranes are the major source for the amniotic fluid before 18 weeks, followed by skin. Therefore the fetal anomalies do not affect the amniotic fluid volume prior to this period. After that period the major source of amniotic fluid is fetal urine followed by the secretions of lungs and membranes. The fetus swallows the amniotic fluid which is absorbed by the fetal intestine. The amniotic fluid homeostasis is maintained by the balance between the inflow from fetal urinary tract and lung secretion and outflow by swallowing and absorption by the fetal GIT. Hence anomalies of the urinary tract and GIT are associated with changes in the amniotic fluid volume – either as polyhydramnios when there is on obstructive anomaly of the GIT or oligohydramnios in anomalies of urinary tract. The degree of polyhydramnios in obstructive anomaly of the GIT decreases with the more distal the obstruction and increases with the gestation as swallowing and peristalsis become more frequent and vigorous.
Esophagus
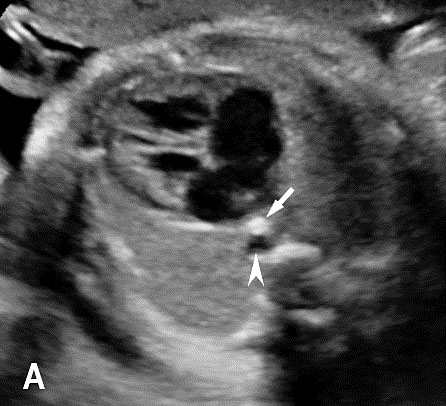
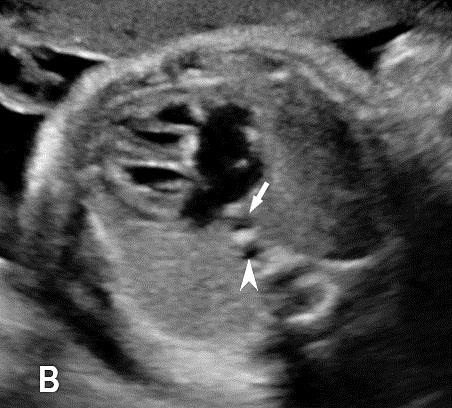
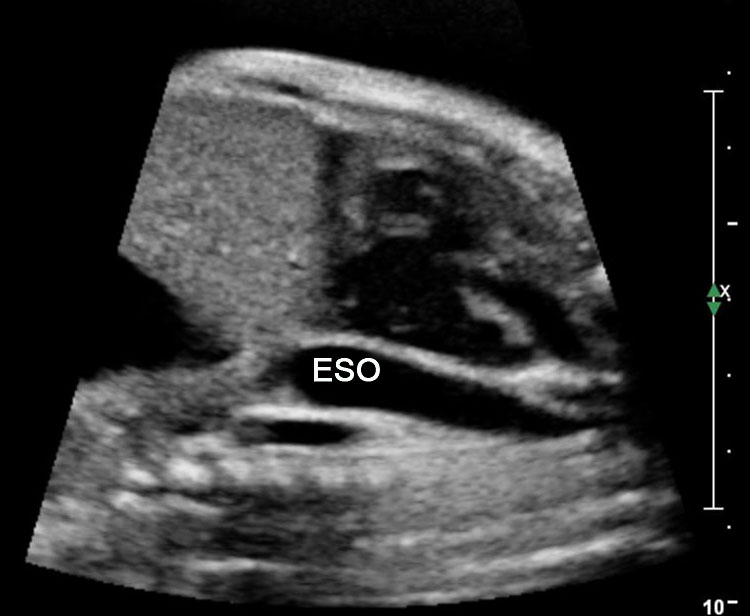
The normal esophagus is a collapsed structure that is not routinely imaged in the fetus as per guidelines, but it can be seen in the second and third trimester. Its appearance varies depending on its status, whether collapsed or fluid filled. The esophagus is seen as a small round echogenic structure or a small round echo-poor structure, between the heart and the descending thoracic aorta in the axial section of fetal thorax at the level of four-chamber view of the heart (Figure 5). On the longitudinal scan collapsed esophagus is seen as a linear triple-line structure (Figure 6A). Occasionally it may be seen as a fluid-filled tube (Figure 6B). In real-time imaging, the peristalsis can also be seen (Video 1). Visualization of the whole length of the esophagus, when intact, is time consuming and requires experience. Furthermore, scanning the esophagus on routine anomaly scans is not mandatory in international guidelines.4 The presence of a patent and normally functioning esophagus is usually inferred by noting fluid in the fetal stomach, so direct visualization of esophagus is unnecessary.
(A) |
(B) |
5
Axial section of fetal chest at the level of four-chamber view showing the collapsed echogenic (A) and round fluid-filled (B) esophagus (arrow) between the heart and descending aorta (arrowhead).
(A) |
(B) |
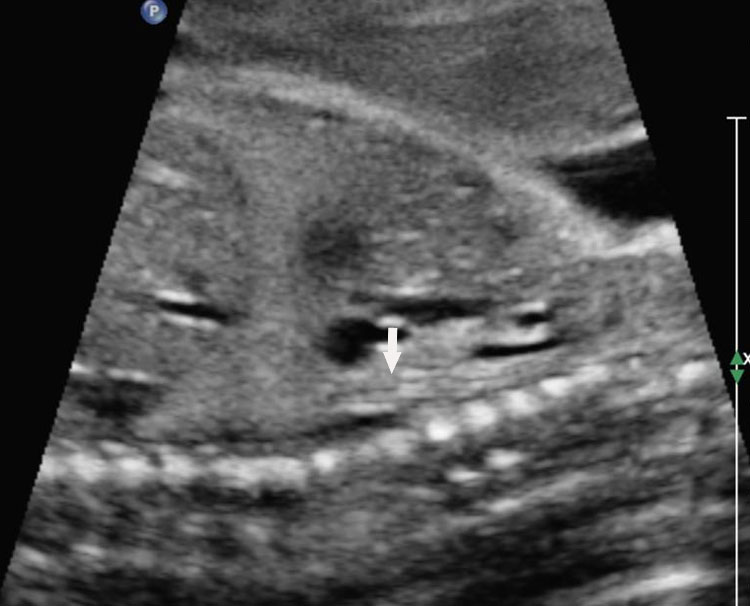
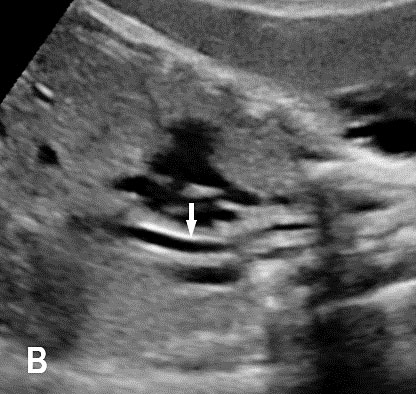
6
Longitudinal section of the fetal chest showing the collapsed triple line (A) and fluid-distended (B) esophagus (arrow).
1
Longitudinal section of fetal chest showing the peristalsis of normal esophagus.
Visualization of stomach by sonography
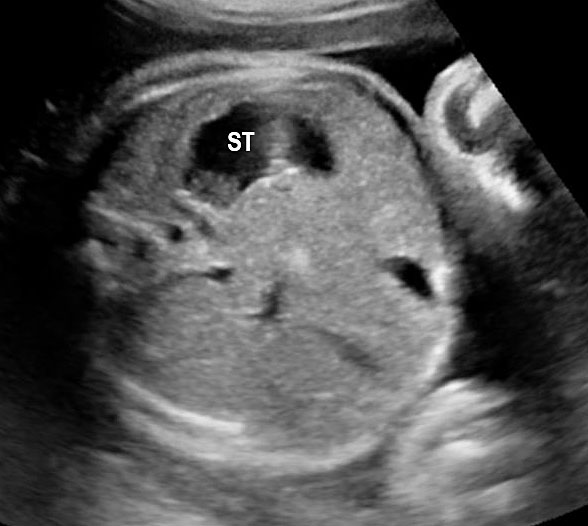
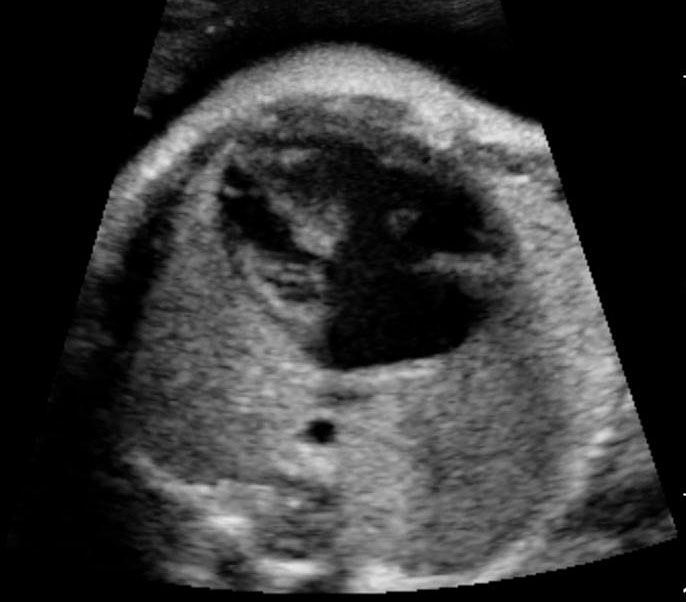
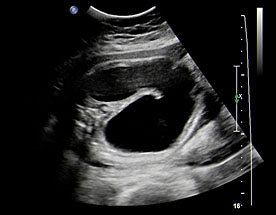
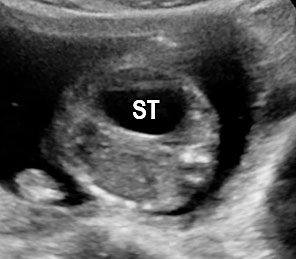
Fetal swallowing starts by 11 weeks of gestation and the fetal stomach, as a “stomach bubble”, is seen as an oval or round echo-poor structure in left upper quadrant of abdomen from 12 weeks onwards (Figure 7). There is marked variation in the size of fetal stomach among fetuses during second and third trimesters due to the fetal swallowing and gastric emptying over time and so the variation in the size of stomach does not carry any significance. If the stomach is not seen in the abdominal circumference (AC) section, then the stomach should be checked for, either on the right side of abdomen or in the chest. If it is still not seen, then the scan should be repeated after 30 minutes.
(A) |
(B) |
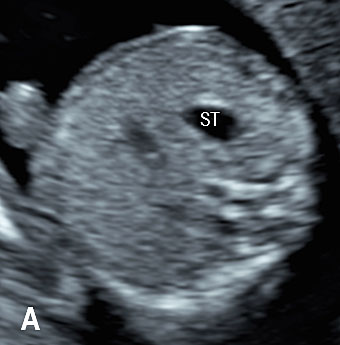
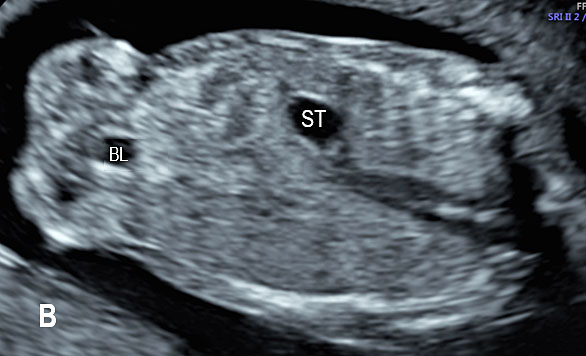
7
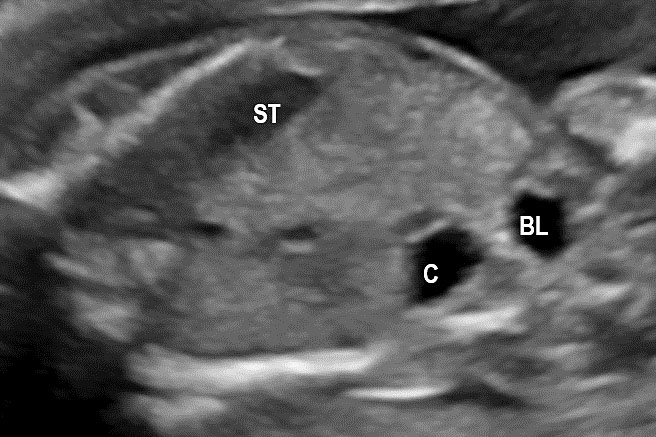
(A) Axial and (B) coronal scan of fetal abdomen at 12 weeks of gestation showing the stomach bubble (ST). BL, urinary bladder.
Esophageal atresia
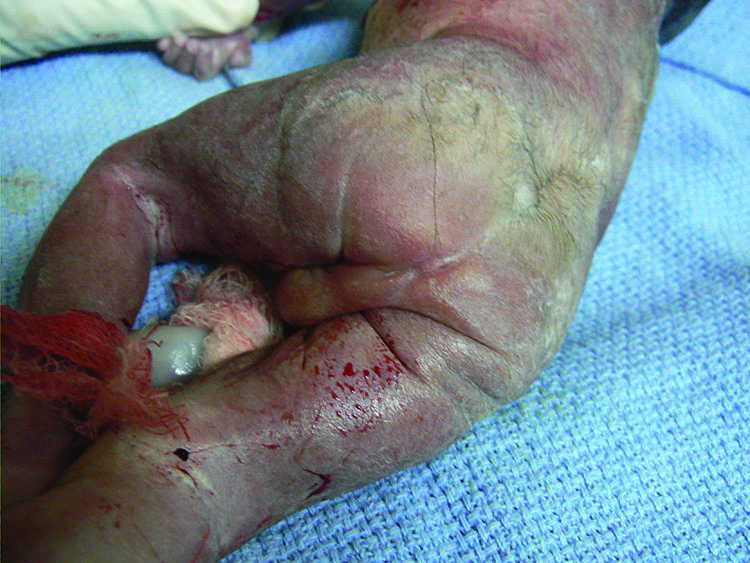
Esophageal atresia (EA) comprises a spectrum of congenital anomalies characterized by a lack of continuity of the esophagus (atresia) with or without the presence of one or more abnormal connections with the trachea (tracheoesophageal fistula). The incidence is 1 in 2500 to 4000 live births with a male preponderance. Even though the survival rate in EA cases has increased to 91–98% in the last decade, mortality remains primarily related to associated anomalies and genetic syndromes. Therefore, prenatal suspicion of EA is important. Increased prenatal detection of EA prompts careful assessment for associated anomalies, provides the opportunity for effective prenatal counseling and has been shown to result in fewer postnatal transfers.5 There are five types of esophageal atresia (Figure 8). The commonest type is esophageal atresia with tracheoesophageal fistula (TEF) (type C) between the trachea and distal segment of esophagus forming 85% of the cases.
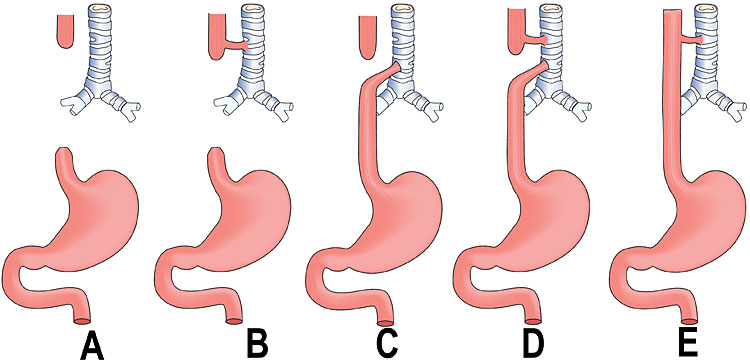
8
Schematic diagram showing the types of esophageal atresia.
Sonographic features
Prenatal diagnosis of EA is still challenging. The main signs detected on routine prenatal ultrasound are polyhydramnios, a small or absent stomach bubble, and a blind‐ending dilated upper esophageal pouch (“pouch sign”).6,7,8,9,10 The use of ultrasound for the prenatal diagnosis of EA is limited by the fact that these signs are operator‐dependent, can be nonspecific and transient. In esophageal atresia without TEF, the fetal stomach is persistently nonvisualized (Figure 9). The scan has to be repeated after an interval to confirm that the stomach is persistently nonvisualized. There is polyhydramnios, which is not attributable to any other anomaly. These features are diagnostic of esophageal atresia without tracheoesophageal fistula. Typical sonographic findings of EA, such as polyhydramnios with an absent stomach, are observed in only 25.9% of EA cases. Normal sonographic findings including normal AFI and stomach shape is seen in 22.2% of EA cases. Polyhydramnios was present in just over half of confirmed cases (56.3%) of EA. This may be influenced by the timing of the prenatal ultrasound, as fetal swallowing is not established until the 5th month of gestation, and polyhydramnios is rarely diagnosed earlier than 24 weeks.7 AFI showed an increasing trend with advancing gestation along with prominent polyhydramnios after 28 weeks. There are other conditions where the stomach bubble is not seen which have to be ruled out before diagnosing the esophageal atresia. Anomalies causing impaired fetal swallowing like cleft lip/palate, masses in oral cavity, neck or chest, esophageal compression due to vascular ring or masses and neuromuscular causes such as anencephaly and fetal akinesia deformation sequence may have nonvisualized stomach bubble. The fetal stomach bubble is also absent in cases of severe oligohydramnios. A small or absent stomach was identified in half of cases of confirmed EA (Figure 10). This sign is subjective, with no consensus at present with respect to the classification of a “small” stomach. It is also difficult to determine, as visualization of the stomach bubble may be transient due to the intermittent nature of fetal swallowing. Even in the absence of a fistula, the stomach may still be visualized as a result of secretions from the gastric mucosa. The combination of polyhydramnios and a small or absent stomach bubble has been shown to increase the accuracy of prenatal diagnosis of EA. Both polyhydramnios and a small or absent stomach bubble have been found to be associated with a higher incidence of false‐positive diagnosis of EA in the context of associated anomalies.8,9 Splenic flexure of colon can mimic a small stomach bubble which can be differentiated in a coronal scan (Figure 11). Anomaly scans do not incorporate gastric-bubble measurements.4
(A) |
(B) |
(C) | |
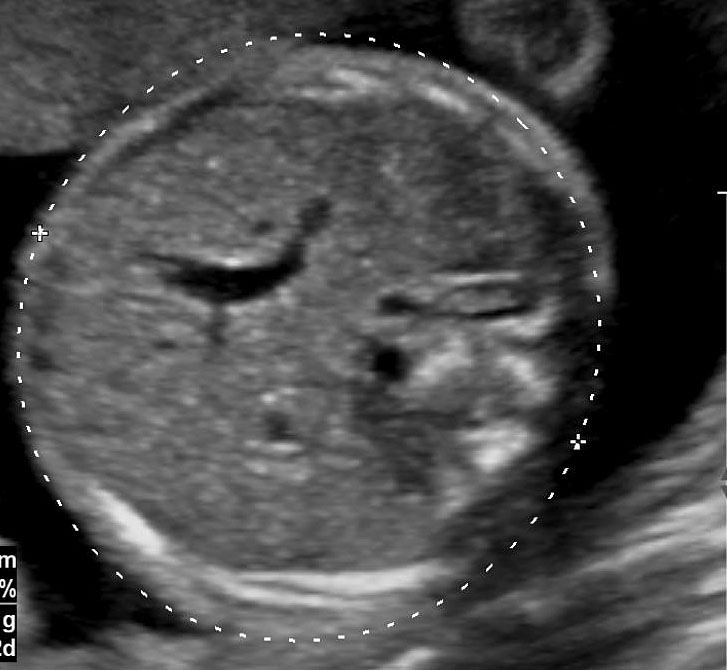
9
(A) Axial and (B) coronal section of upper abdomen in a fetus with esophageal atresia without tracheoesophageal fistula showing the nonvisualized stomach bubble with (C) polyhydramnios. GB, gall bladder; BL, urinary bladder.
(A) |
(B) |
(C) |
(D) |
10
(A) Axial and (B) coronal section of upper abdomen in a fetus with esophageal atresia without tracheoesophageal fistula showing the small stomach bubble (arrow) with polyhydramnios. Coronal scan of fetal neck showing the fluid distended blind esophageal pouch (arrow) in (C) and collapsed pouch in (D). GB, gall bladder; BL, urinary bladder; UPV, umbilicoportal vein.
(A) |
(B) |
(C) | |
11
(A) Axial scan of fetus with esophageal atresia without tracheoesophageal fistula with polyhydramnios (B) showing splenic flexure of colon mimicking a small stomach bubble (arrow). Coronal scan (C) confirms that it is colon (arrows).
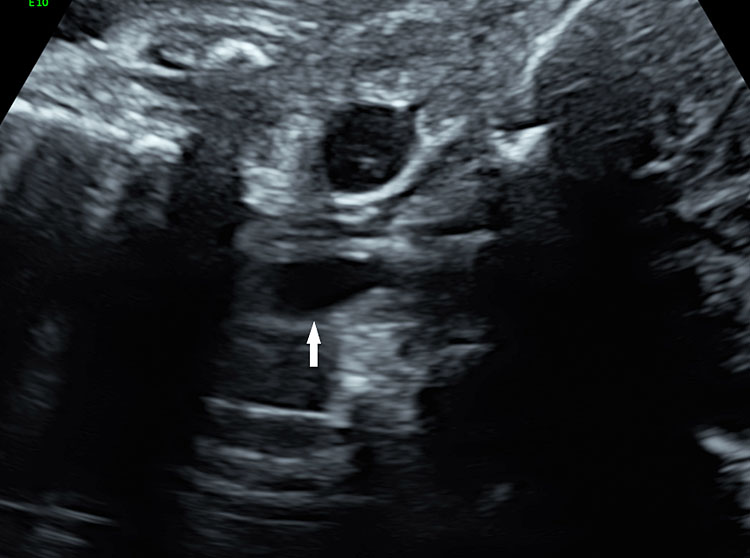
In the presence of polyhydramnios the visualization of stomach bubble does not rule out esophageal atresia since the commonest type is with tracheoesophageal fistula (85%) through which amniotic fluid reaches the stomach (Figure 12A). This type can be diagnosed by looking for the blind ending proximal esophagus which fills up and empties during fetal swallowing. This is called as pouch sign (Figure 12B,C and Video 2). The pouch sign has been proposed as the most reliable sign for the diagnosis of EA.6,10 To see the pouch sign a coronal scan of the neck is performed and the transducer is rocked between the spine and the trachea (Figure 13 and Video 3). The blind esophageal pouch can be seen as a transient finding when it fills up during fetal swallowing (Video 3). If it is persistent for some time, the pouch can be imaged in transverse section also (Figure 12D). The pouch sign is seen in esophageal atresia without TEF also (Figure 10C). However, it is not necessary to look for this sign when the features are characteristic, namely, persistently nonvisualized stomach associated with polyhydramnios, which is not attributable to any other anomaly. The pouch sign will be useful in esophageal atresia without TEF when a small stomach bubble is seen (Figure 10).
(A) |
(B) |
(C) |
(D) |
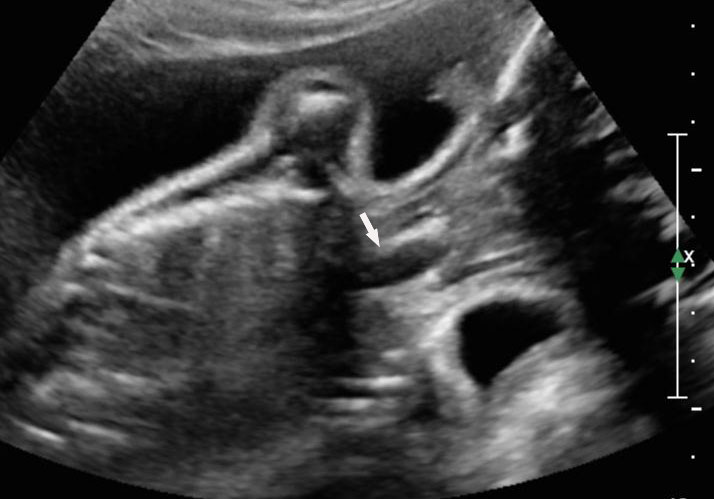
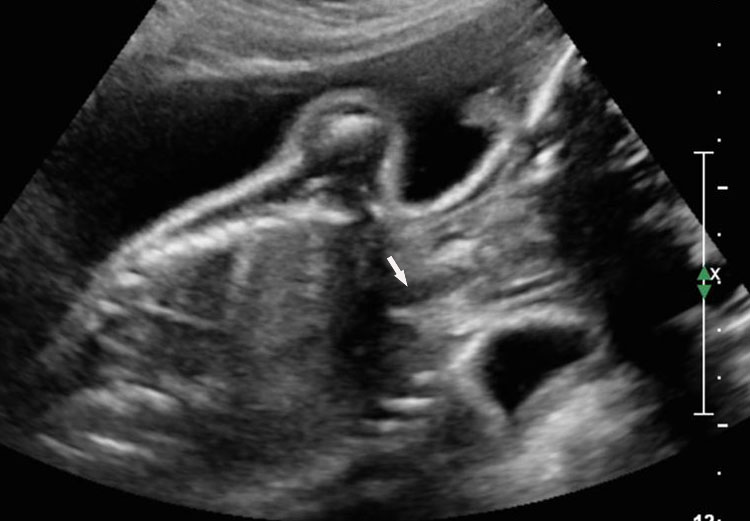
12
Esophageal atresia with tracheoesophageal fistula. (A) Axial section a fetal upper abdomen showing the normal stomach bubble (ST). (B) Coronal scan of fetal neck showing the fluid-distended blind esophageal pouch (arrows). (C) Collapsed pouch (Video 3). (D) Axial scan of neck showing the round fluid distended pouch.
2
Technique of seeing pouch sign: coronal scan of neck is obtained and the probe is rocked between spine and trachea.
(A) |
(B) |
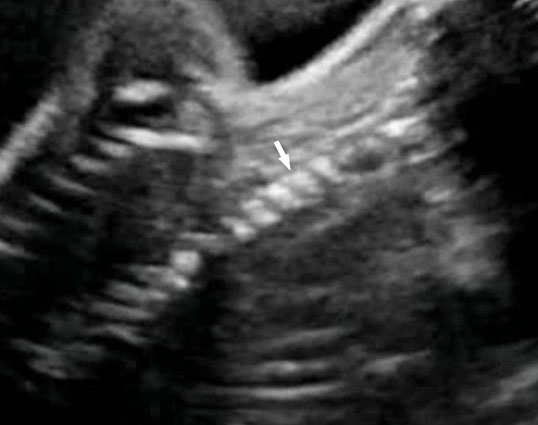
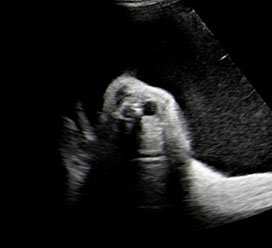
13
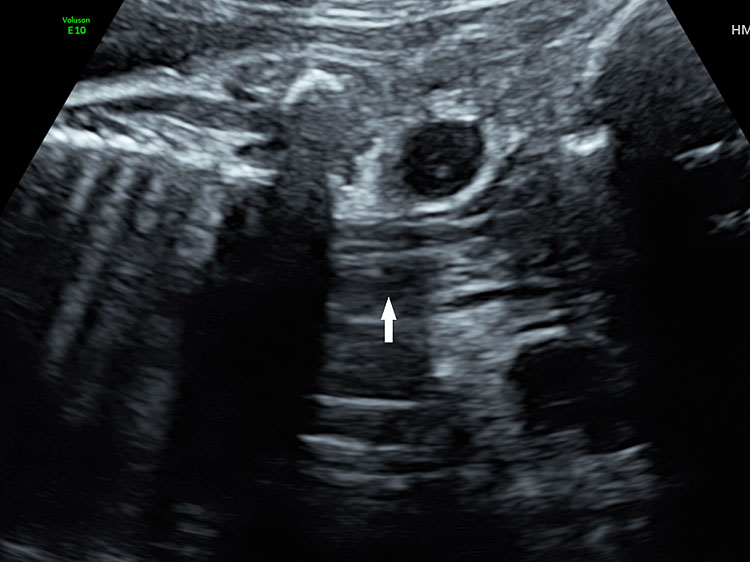
Coronal scan of fetal neck showing the fluid filled trachea (arrow) in (A) anteriorly and cervical spine (arrow) in (B) posteriorly (Video 2).
3
Pouch sign: coronal scan of fetal neck showing the filling and collapsing of the blind oesophageal pouch.
Features to differentiate trachea and esophageal pouch are shown in Table 1, Figure 14 and Video 4.
1
Features to differentiate trachea and esophageal pouch
Trachea | Esophagus – Pouch |
Bifurcates low down | Does not bifurcate |
Remain stationary | Empties and fills up |
Cylindrical in shape | Oval in shape |
Color Doppler: during fetal breathing, color change is noted indicating fluid movement | |
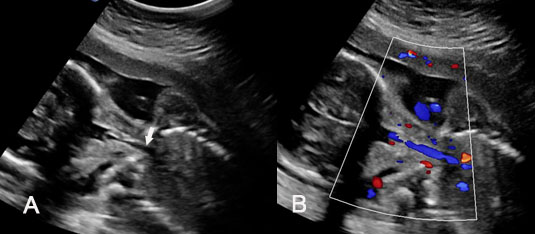
14
(A) Gray scale and (B) color Doppler images of coronal scan of neck showing the fluid-filled cylindrical trachea filling with color due to fetal respiration (Video 4).
4
Coronal gray scale and color Doppler study of fetal neck showing the fluid filled trachea which fills up with color during fetal breathing.
Very rarely the pouch sign may be seen in esophageal obstruction due to a vascular ring such as double aortic arch (Figure 15).
(A) |
(B) |
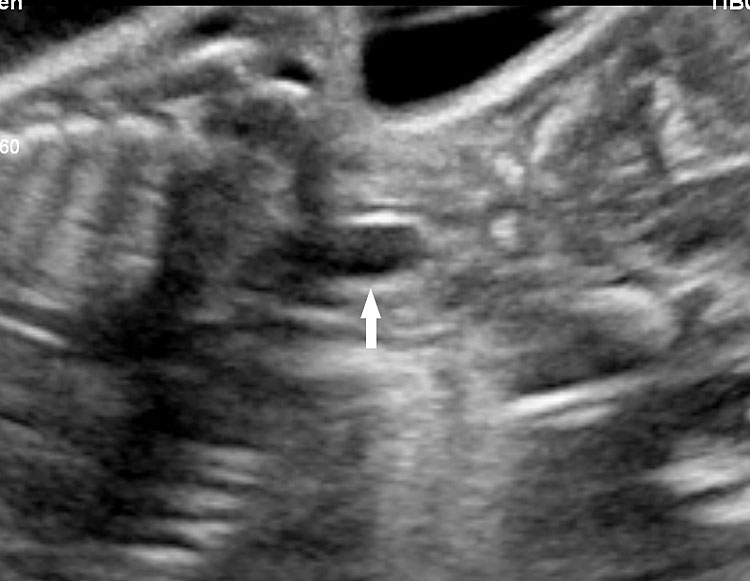
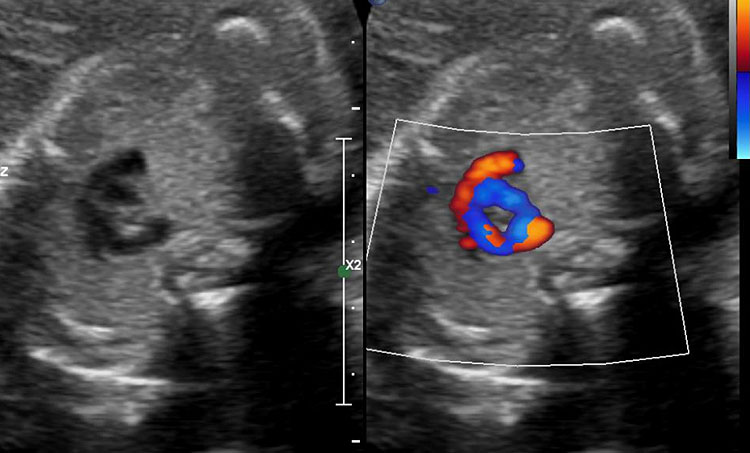
15
(A) Coronal scan of fetal neck showing the fluid distended blind esophageal pouch (arrow). (B) Axial scan of upper chest demonstrating the trachea within the vascular ring formed by double aortic arch.
On the routine mid-trimester scan, the suspicion rate (small/absent stomach and/or worsening polyhydramnios) was 19.4% and the detection rate (esophageal pouch) was 4.3%. On the third-trimester anomaly scan, 43.9% of cases were suspected (either small/absent stomach or worsening polyhydramnios) and 33.9% were detected. Just over 30% (23/75) of cases with EA/TEF did not display any sonographic signs and therefore could not have been suspected prenatally. Prenatal detection of EA/TEF is not feasible before the late second trimester.4 Fetal MRI has the highest sensitivity (94.7%) and therefore appears to be an important non‐invasive tool for improving the prenatal diagnostic accuracy of EA.
Esophageal atresia can be isolated or associated with other anomalies. EA poses a high risk of associated anomalies, such as vertebral, gastrointestinal, cardiovascular, renal, or limb abnormalities, in 50% cases, and 10% EA cases are diagnosed with VACTERL (vertebrae, anus, cardiac, tracheoesophageal, renal and limb) association (Figure 16). Chromosomal anomalies are also common with a prevalence of 5–10%, and most of them are trisomies. Prognosis is good in isolated esophageal atresia.11
(A) |
(B) |
(C) | |
16
Esophageal atresia in VACTERL association. (A) Pouch sign. (B) Unilateral renal agenesis with lying down adrenal sign (arrow). (C) One upper limb showing radial ray aplasia.
Gastric pseudomass
Gastric pseudomass is defined as a soft tissue mass within the stomach (Figures 17 and 18A). It is not a neoplasm. It has a variable sonographic appearance and may be mobile. The pseudomass is produced by aggregates of swallowed fetal cells denuded from the fetal skin. Usually it is a transient finding. There is increased incidence associated with bowel obstruction and ventral wall defects.
|
|
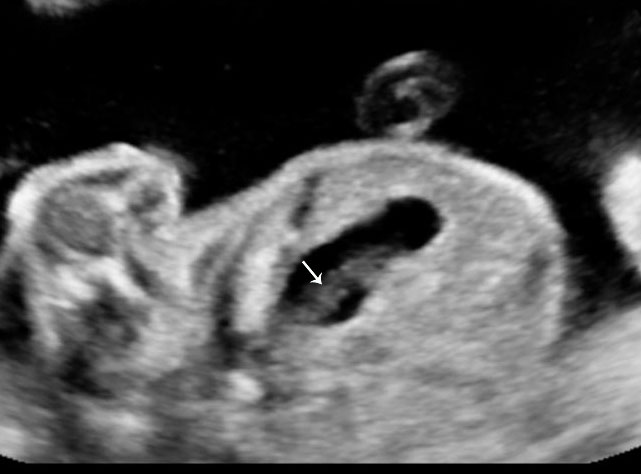
17
Gastric pseudomass (arrow) in two fetuses.
(A) |
(B) |
(C) |
(D) |
(E) |
(F) |
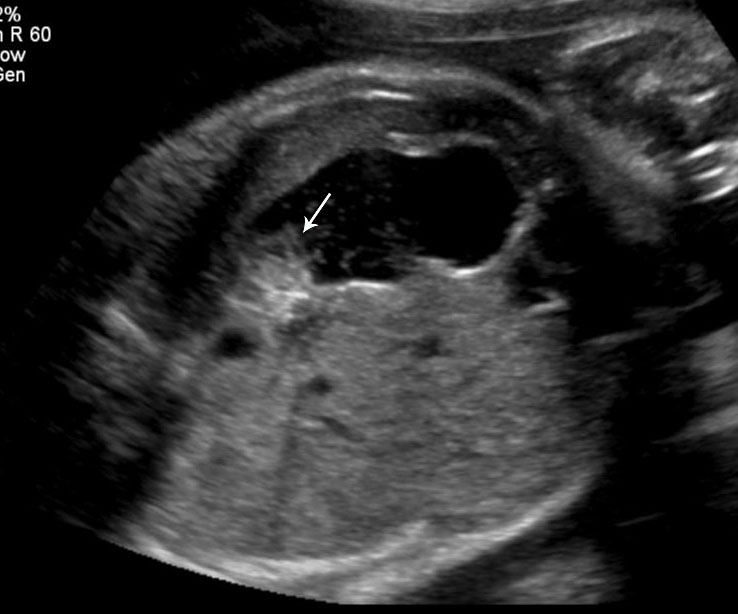
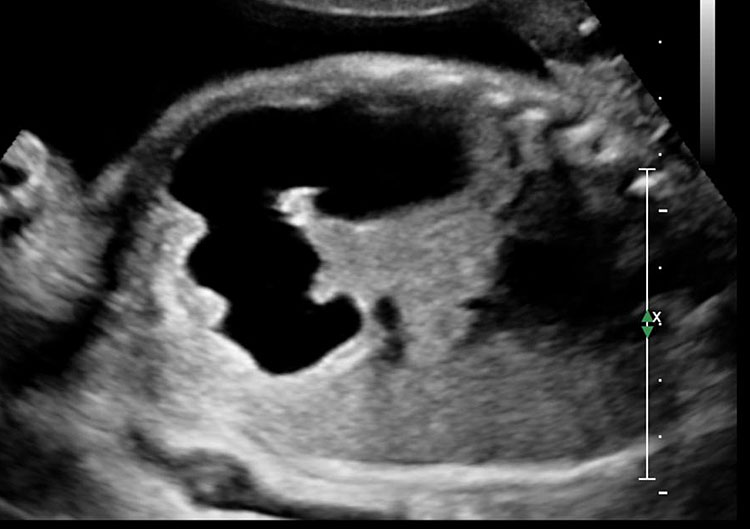
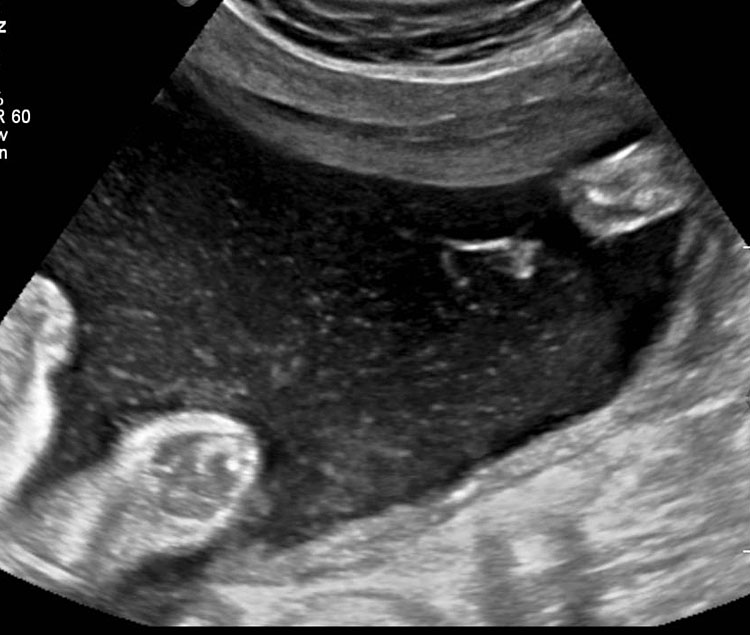
18
Pyloric atresia. (A) Axial and (B) coronal scan showing the dilated stomach upto pylorus with pseudomass (arrow). (C) Snow flake sign in amniotic fluid (Video 5). (D) Sagittal scan of fetal chest showing the fluid distended esophagus. (E) Four-chamber view of heart showing AVSD. (F) Coronal scan of fetal chest and upper abdomen shows the dilated esophagus (arrow) and stomach (ST).
5
Snow flake sign: scan of an amniotic fluid pocket shows the movement of debris in it.
Pyloric atresia
Pyloric atresia (PA) is a very rare anomaly with an incidence of about 1 in 100,000 live births. It is seen in about 1% of all intestinal atresia. PA may occur as an isolated anomaly (35%), or associated with epidermolysis bullosa (EB) (40%) or other abnormalities (25–55%), such as multiple intestinal atresia, renal and urethral abnormalities, cardiac malformations, aplasia cutis, onychodystrophy, and gallbladder agenesis.12 Major association is with epidermolysis bullosa (PA–EB). Epidermolysis bullosa is an autosomal recessive condition affecting the skin, manifesting as blisters, erosions and scars. Defect in ITGA6, ITGB 4 and PLEC genes have been indicated. It has an incidence of 1 in 100,000 live births.
Sonographic features of pyloric atresia are polyhydramnios, grossly dilated stomach with hyperperistalsis and dilated esophagus due to regurgitation caused by the distal obstruction (Figure 18). There will be no double bubble sign. There may be pseudo-double bubble sign due to dilated stomach which is to be carefully differentiated. In PA-EB syndrome, snowflake sign of echogenic debris in amniotic fluid due to excess skin denudation may be seen in real-time scan (Figure 18C and Video 5).13 There may be pseudomass in the stomach. The sonographic signs are seen usually in late second trimester. Other associated anomalies may be seen, more commonly cardiac and urinary tract anomalies. PA-EB syndrome is a highly lethal condition. Early sonographic diagnosis is possible with family history. Early genetic diagnosis can be offered.
Duodenal obstruction
Congenital duodenal obstruction could be classified as complete congenital duodenal obstruction (CCDO) and incomplete congenital duodenal obstruction (ICDO). Complete duodenal obstruction usually results from duodenal atresia. Incomplete duodenal obstruction can be caused by an intraluminal web with a central opening, annular pancreas or intestinal malrotation. In cases of intestinal malrotation, extrinsic compression through Ladd’s bands causes duodenal obstruction. Incidence of duodenal obstruction is 1 in 5000 to 10,000 live births. Approximately 20–40% of all infants with duodenal atresia have Down syndrome and 8% of infants with Down syndrome have DA.14
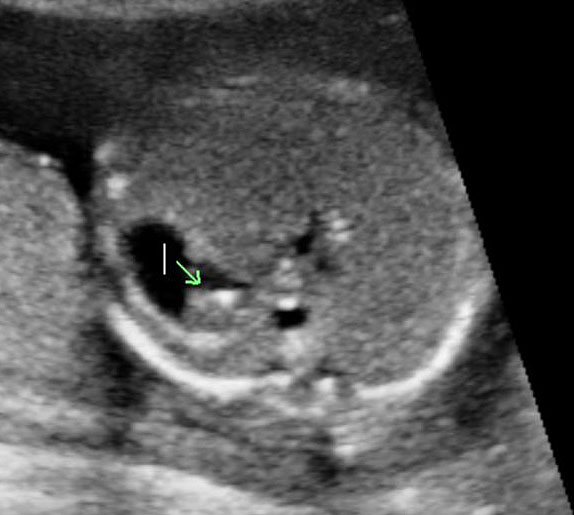
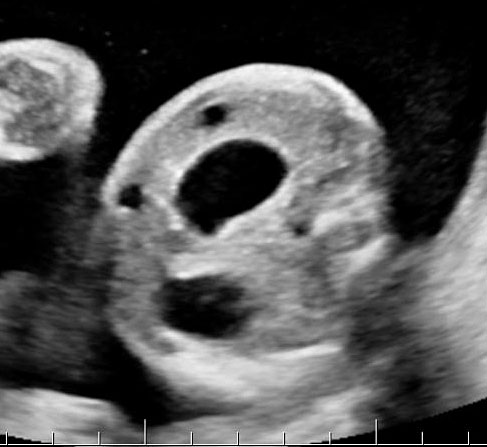
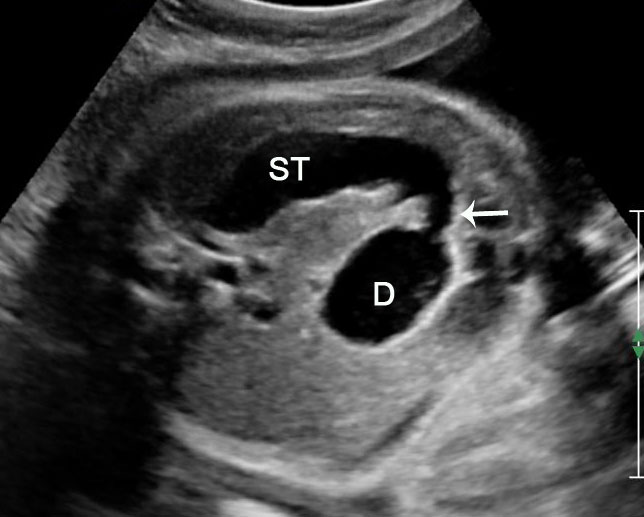
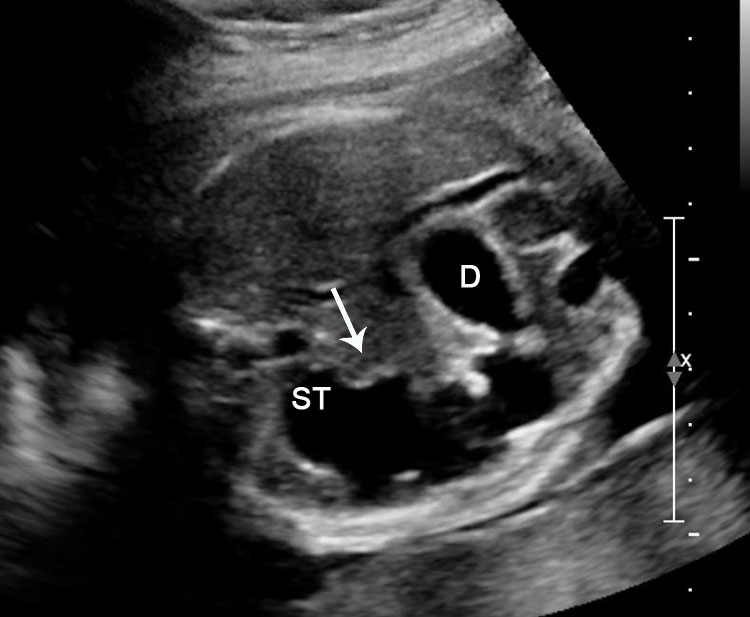
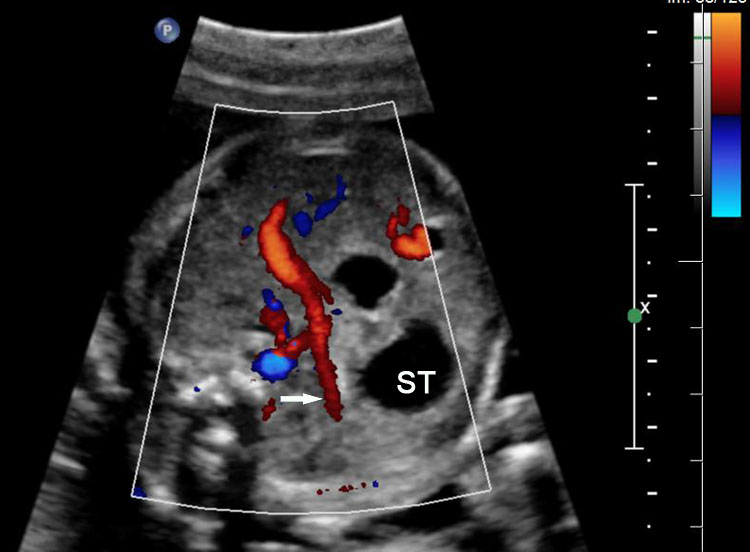
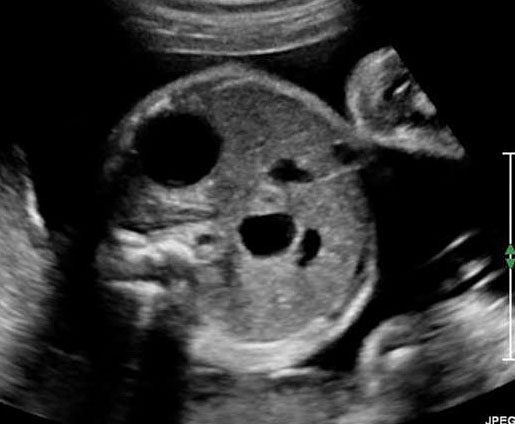
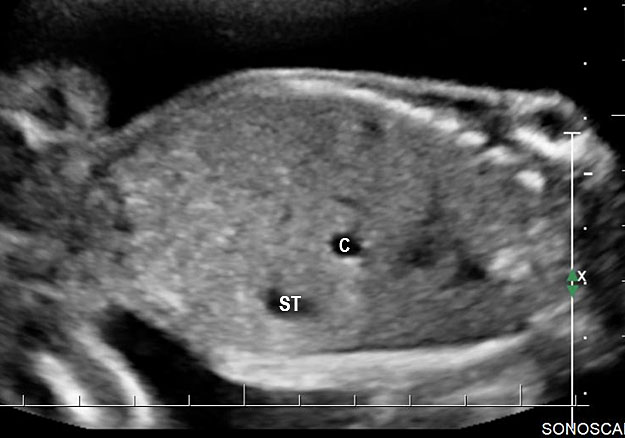
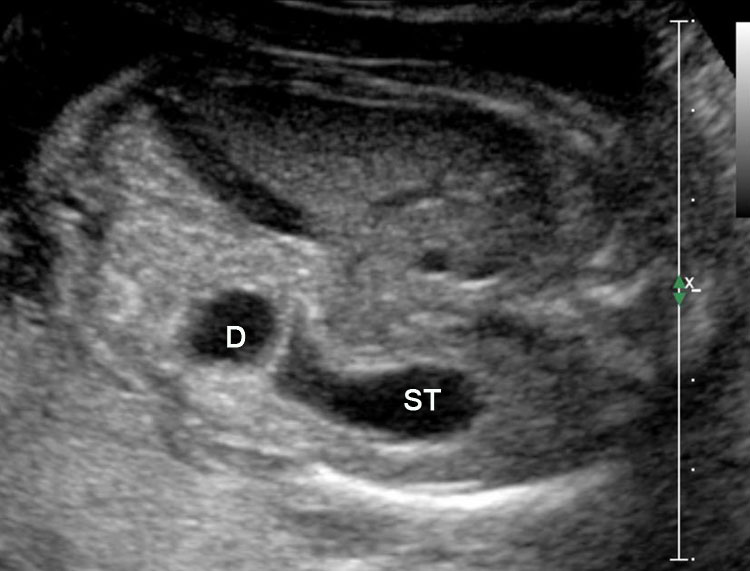
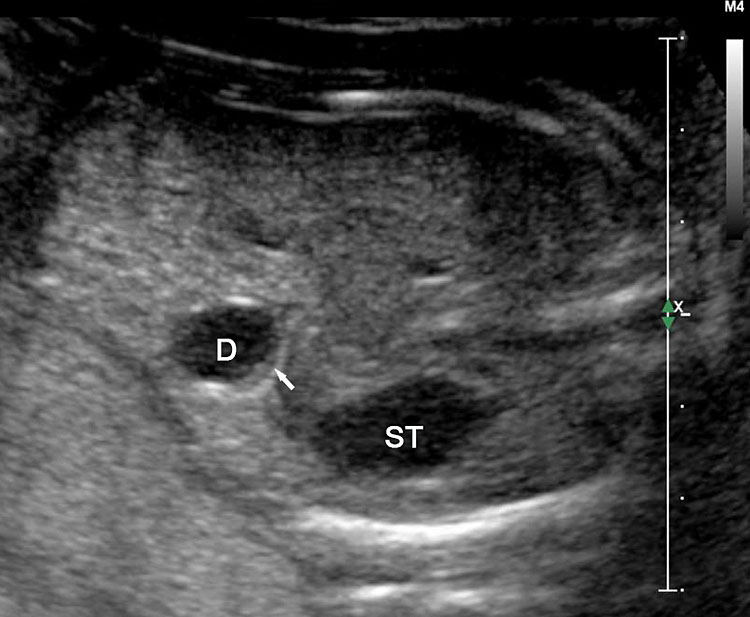
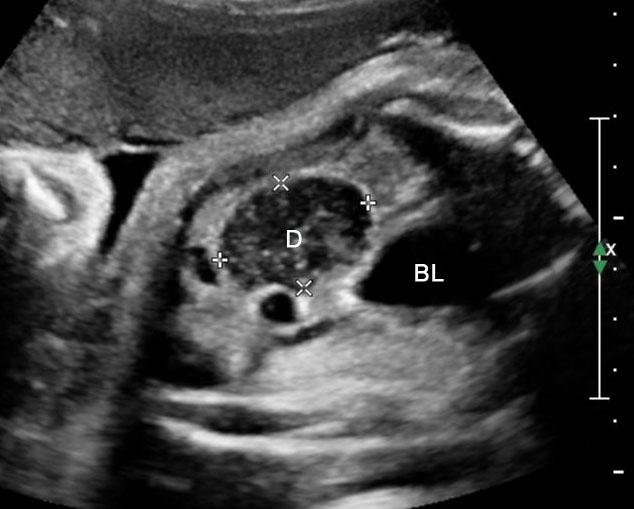
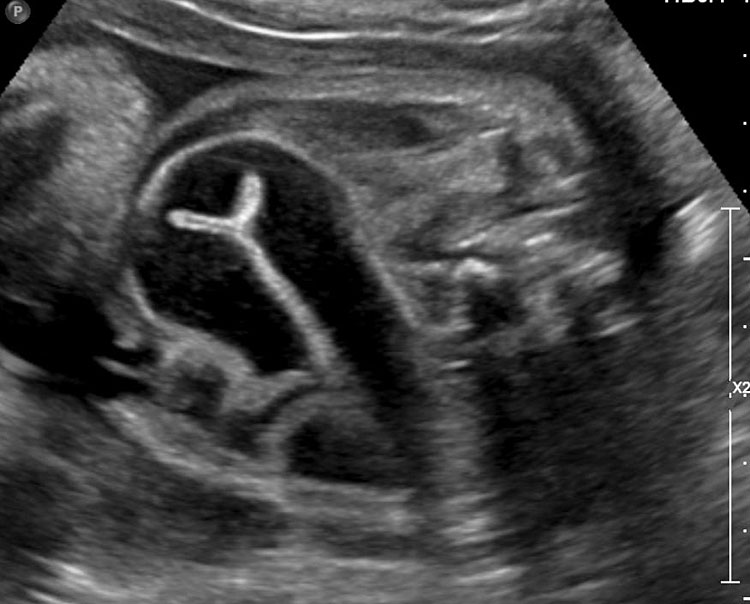
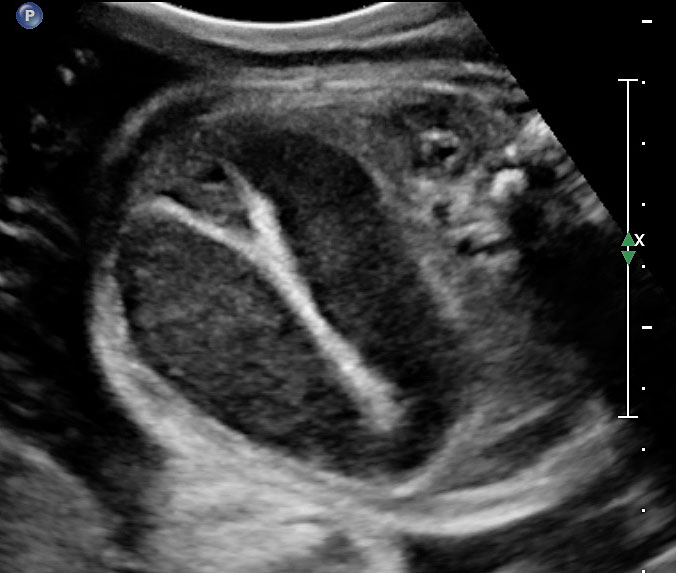
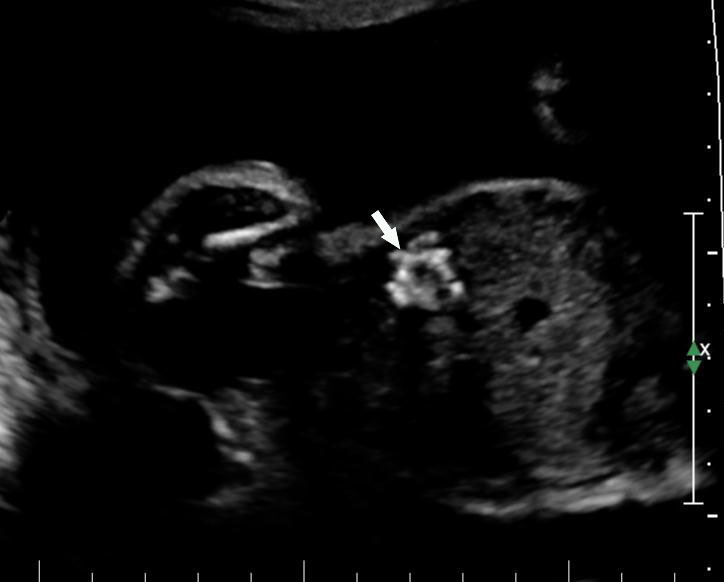
Sonographic sign of duodenal obstruction is “double bubble” sign in the axial section of fetal upper abdomen, with polyhydramnios (Figure 19A). The two bubbles are on either side of midline. The fluid-distended stomach will cross the midline to the dilated duodenum. The continuity between the two bubbles through narrow pylorus should be demonstrated (Figure 19B). Gastric hyperperistalsis will be seen (Figure 19C and Video 6). If obstruction is partial, dilatation of the stomach and duodenum is less marked with delayed manifestation. It happens in fenestrated duodenal web, malrotation of midgut due to Ladd’s bands or annular pancreas. In case of malrotation of midgut, color Doppler study will show inversion of relationship of superior mesenteric artery and vein, where the vein is seen to the left of artery (Figure 20). Bishop et al. reported a mean gestational age for positive ultrasound suggestive of duodenal atresia of 29 + 5/7 weeks (ranging from 19 + 6/7 to 38 + 4/7).15 There are sparse data on the criteria for prenatal detection of CDO. Apart from the double bubble sign, polyhydramnios can be a sign of high bowel atresia due to the lack of re-absorption of amniotic fluid.16 Maternal polyhydramnios is thought to serve as an aid in the diagnosis of duodenal obstruction;17 however, the presence of polyhydramnios varies greatly in the literature (from 32% to 81%).14 Bittencourt and colleagues reported an overall prenatal detection rate of 44% by findings of polyhydramnios and double bubble sign on antenatal ultrasound exams.18 Overall, there is very little information on the detection rate of duodenal obstruction in routine ultrasound. Furthermore, even fewer data are available on the prenatal diagnosis of ICDO.19
Prognosis of DA depends on associated structural and chromosomal abnormalities. Since there is a high risk of Down syndrome it is mandatory to offer prenatal genetic counseling and invasive testing. The risk of Down syndrome increases further if there is associated atrioventricular septal defect (AVSD).
(A) |
(B) |
(C) | |
19
Duodenal obstruction. Axial section of fetal upper abdomen showing the double bubble sign of dilated stomach and duodenum on either side of midline (A) with continuity of the two bubbles through the narrow pylorus (arrow) in (B) and hyperperistalsis of stomach seen as multiple constrictions (arrow) in (C) and also in Video 6. ST, stomach; D, duodenum.
6
Gastric hyperperistalsis in duodenal obstruction.
(A) |
(B) |
(C) |
(D) |
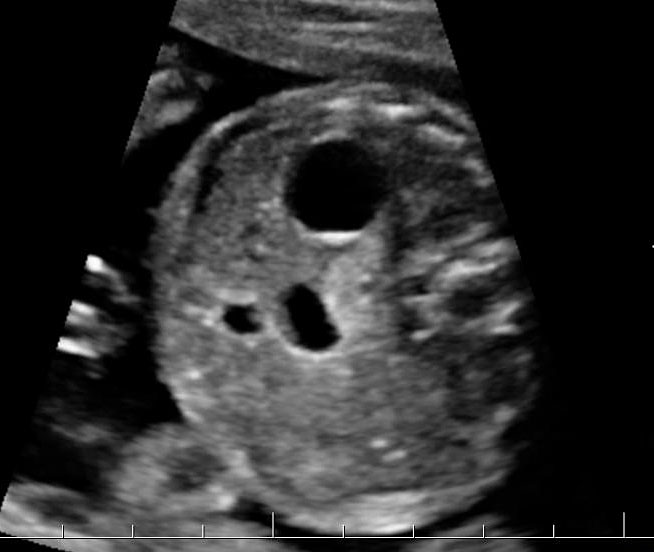
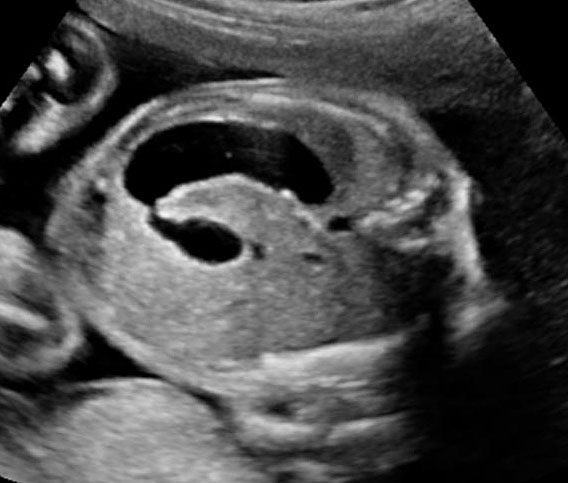
20
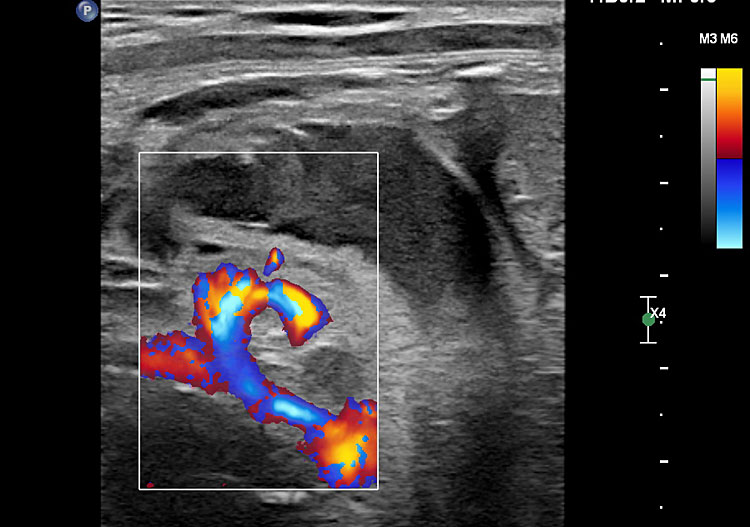
Duodenal obstruction due to malrotation of midgut. Axial sections of a 30 weeks' fetus showing the double bubble sign (A) and continuity of two bubbles (B). Color Doppler image (C) through liver showing the splenic vein (arrow) extending from left side, posterior to stomach extending to the right side as the portal vein with in the liver. (D) Oblique section through mid-abdomen showing the inversion of relation-ship of superior mesenteric artery and vein with vein to the left of artery indicating malrotation.
Differential diagnosis
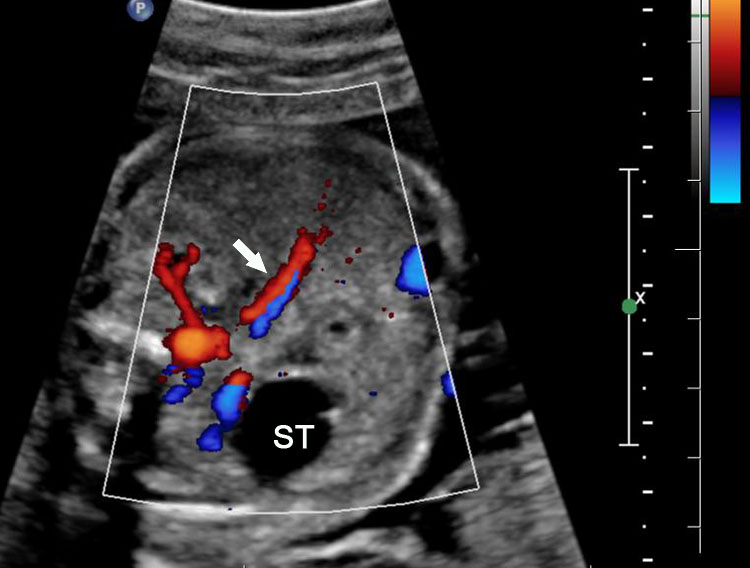
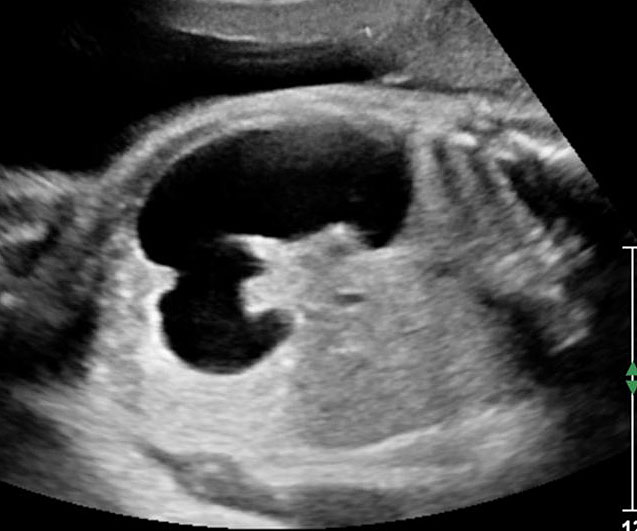
When stomach appears well distended it may mimic double bubble sign (pseudo double bubble sign), but the narrow pylorus will not be seen in coronal oblique scan. It will not cross the midline (Figure 21). A cystic mass in upper abdomen like liver/splenic cyst, choledochal cyst, adrenal cyst, dilated gall bladder, renal cyst, etc. may be mistaken for a double bubble sign. Features of continuity and peristalsis will differentiate them.
(A) |
(B) |
(C) | |
21
Pseudo double bubble sign: (A) Axial section showing double bubble and (B) coronal section showing only dilated stomach. Duodenal bubble is not seen. (C) Schematic to explain the pseudodouble sign.
Duodenal and esophageal atresia
Very rarely there can be atresia of both the esophagus and duodenum resulting in closed loop obstruction of stomach. On sonography, the stomach is grossly dilated due to accumulated secretions. There is a large double bubble sign with dilated distal esophagus (Figure 22). Double atresia can manifest in earlier gestation since it is a closed loop obstruction (Figure 23) The risk for Down syndrome is very high in double atresia.
(A) |
(B) |
22
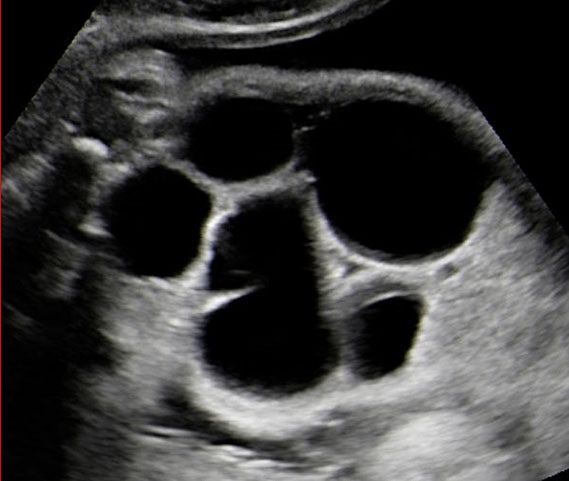
Double atresia of esophagus and duodenum. (A) Axial section showing the giant double bubble sign and (B) the continuity of the bubbles.
(A) |
(B) |
(C) | |
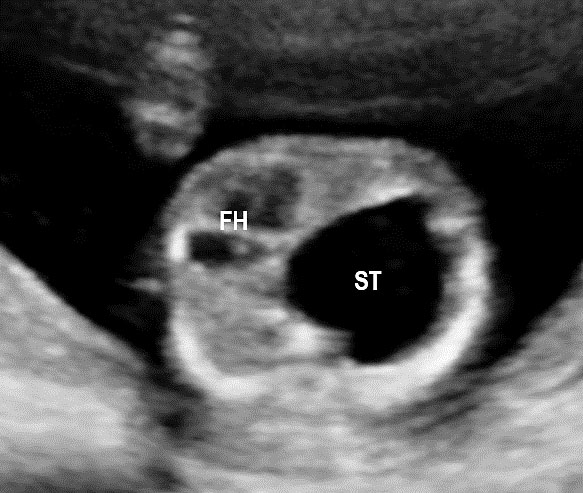
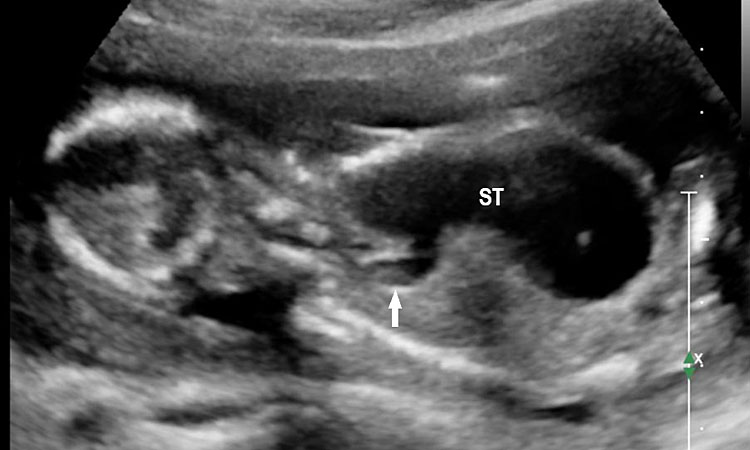
23
Double atresia of esophagus and pylorus at 17 weeks. Axial section of upper abdomen (A) showing the large stomach bubble (ST) and chest (B) demonstrating part of the grossly dilated stomach in lower chest on left side pushing the fetal heart to right (C) Coronal scan of fetal chest and abdomen confirming dilated stomach in chest and abdomen and dilated lower esophagus diagnostic of closed loop obstruction due to double atresia.
Bronchopulmonary foregut malformation
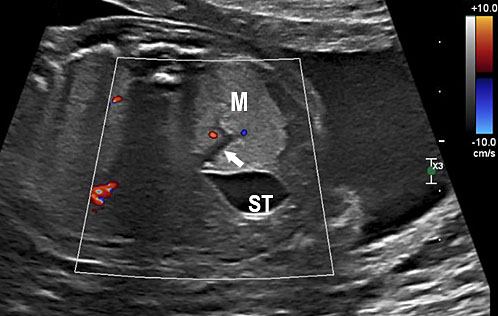
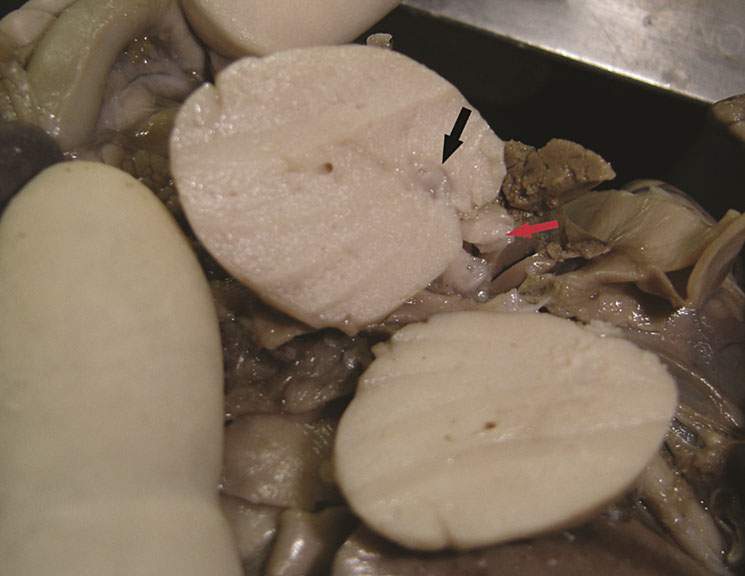
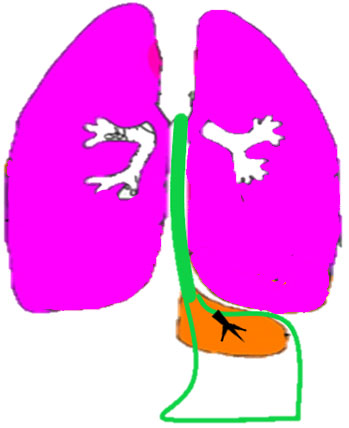
Communicating bronchopulmonary foregut malformation (CBPFM) is a rare congenital malformation involving both the digestive and respiratory systems, characterized by congenital pulmonary sequestration and the formation of an abnormal communication with the digestive tract. The bronchus can communicate with the stomach or esophagus. In the axial section of fetal abdomen when an echogenic mass is seen posterior to the stomach, an effort is made to identify the organ of origin. If spleen is seen separately and left adrenal is imaged normally, then infradiaphragmatic bronchopulmonary sequestration is to be considered. A tubular cystic structure that does not show flow on color Doppler study representing the bronchus can be seen communicating with the stomach (Figure 24). The bronchus communicating with the stomach differentiates this from infradiaphragmatic bronchopulmonary sequestration.
(A) |
(B) |
(C) |
(D) |
(E) | |
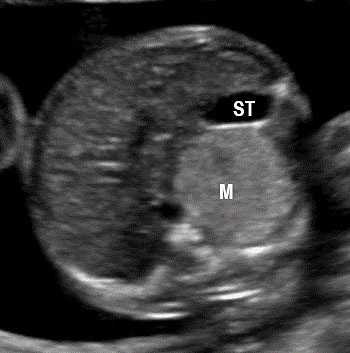
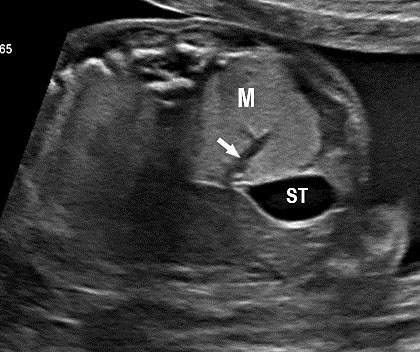
24
Bronchopulmonary foregut malformation. (A) Axial scan of fetal upper abdomen shows intraabdominal echogenic mass (M) posterior to stomach (ST), (B) oblique scan showing a tubular branching fluid filled structure (arrow) within the mass not showing flow on color Doppler in (C) indicating bronchus in in the mass connecting to fundus of stomach, (D) fetal pathology photograph showing the mass with the bronchus (black arrow communicating with the stomach (red arrow), and (E) schematic diagram to explain the condition.
Gastrointestinal duplication cysts
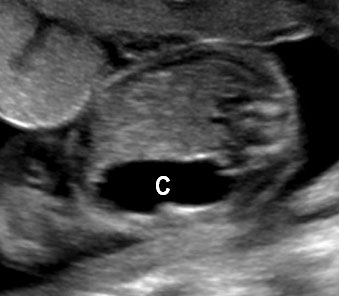
Gastrointestinal duplication cysts are rare congenital malformation of gastrointestinal tract (GIT) representing an embryological failure of recanalization. Incidence is 1 in 4500 live births. They can occur anywhere along the GIT, but are more common in small bowel (80%) followed by esophagus (10%), large bowel and stomach (5%). They share a muscular layer and arterial blood supply with the adjacent bowel. Some have ectopic gastric mucosa. The cysts remain along the mesenteric border of the bowel. They can be cystic (80%) or tubular (20%) and may or may not communicate with the GI tract. The tubular type can vary in length and communicate with native lumen in one or more places. The wall of duplication cysts contains two layers, the mucosal layer and muscle layer, and shares blood supply of the adjacent bowel. The cysts may show peristalsis. They can cause complications of bowel obstruction, volvulus, intussusception and bleeding from heterotopic gastric mucosa. Therefore, antenatal diagnosis is very valuable for surgical excision of the cyst in a planned and timely manner before the complications can develop. Although the differential diagnosis for a fetal abdominal cystic mass is extensive, a systematic approach can narrow the possibilities.
Esophageal duplication cysts
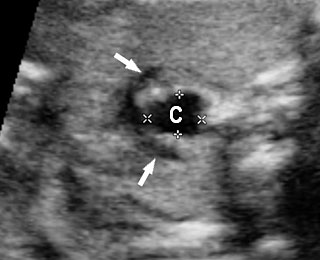
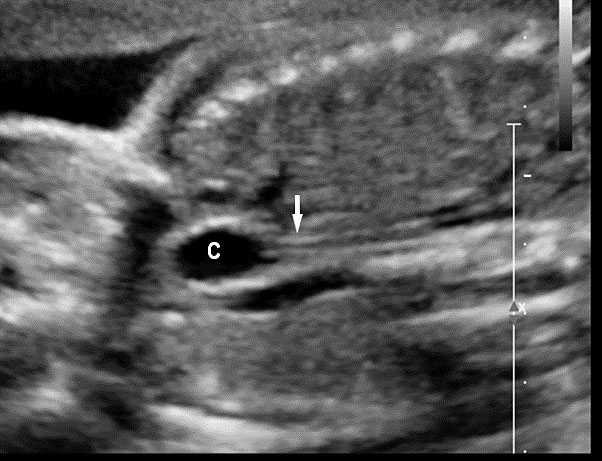
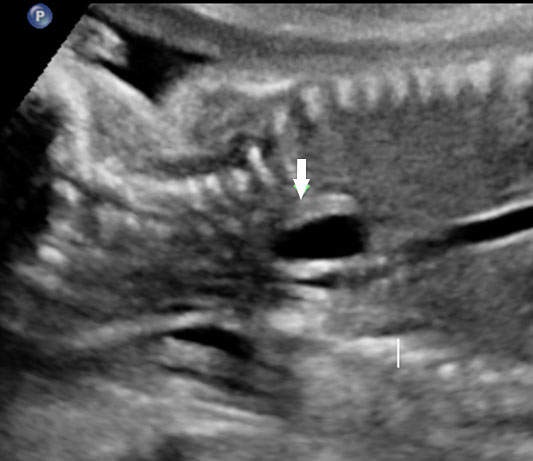
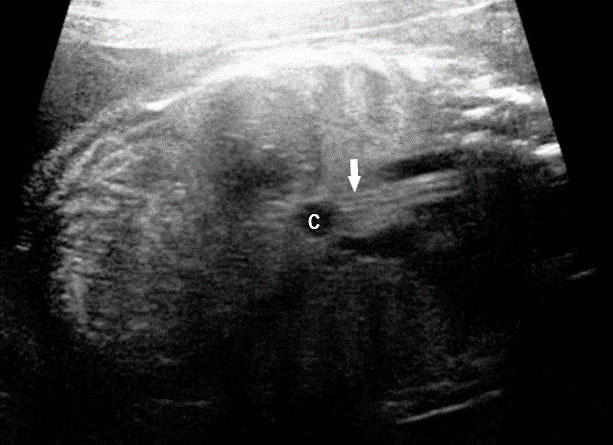
Esophageal duplication cysts can occur anywhere along the esophagus, and are usually asymptomatic. The majority (about 90%) do not communicate with the esophageal lumen. More than half occur in the lower third of the esophagus. Duplication cyst of thoracic esophagus is seen as a cystic mass in midline of fetal chest posterior to the heart in continuity with triple line esophagus. The gut signature in the wall confirms the diagnosis (Figure 25). Associated spinal anomaly is seen in case of a neurenteric cyst. In case of a duplication cyst of lower third of the esophagus, the cyst is seen in the upper abdomen of the fetus just below the diaphragm in close proximity to the fundus of stomach bubble in line with abdominal esophagus. The gut signature in the wall confirms the diagnosis (Figure 26).
(A) |
(B) |
(C) |
(D) |
25
Duplication cyst of thoracic esophagus. (A) Grayscale and (B) color Doppler axial scans of fetal thorax reveals a cyst (C) posterior to the bifurcation of pulmonary artery (arrows). (C) Coronal scan showing the cyst continuous with triple line esophagus (arrow) and gut signature in the wall (arrow) in (D).
(A) |
(B) |
(C) | |
26
Duplication cyst of abdominal esophagus. (A) Axial scan of fetal upper abdomen shows the cyst (C) posteromedial to the stomach bubble (ST). (B) Coronal scan showing the cyst close to midline below the diaphragm. (C) Oblique scan shows the cyst in continuity with the triple line lower esophagus (arrow).
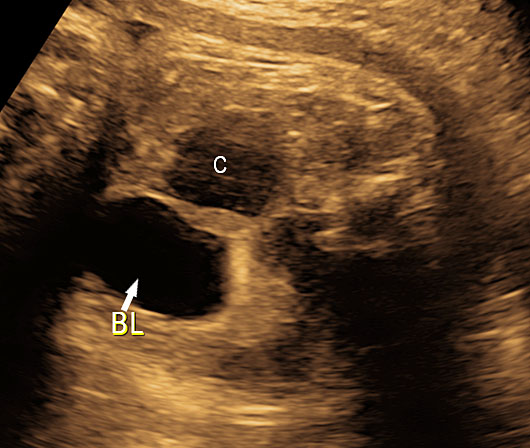
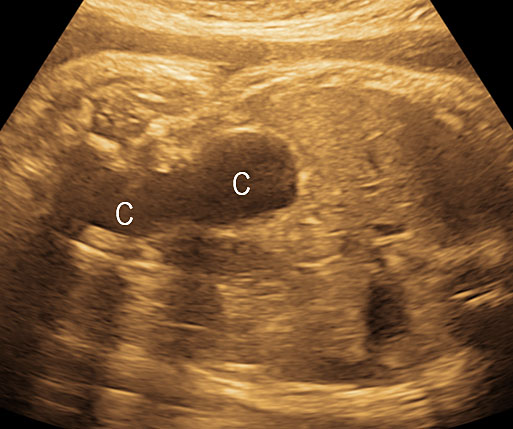
Gastric duplication cysts
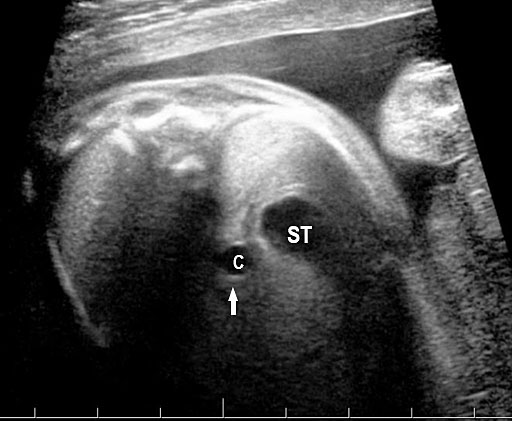
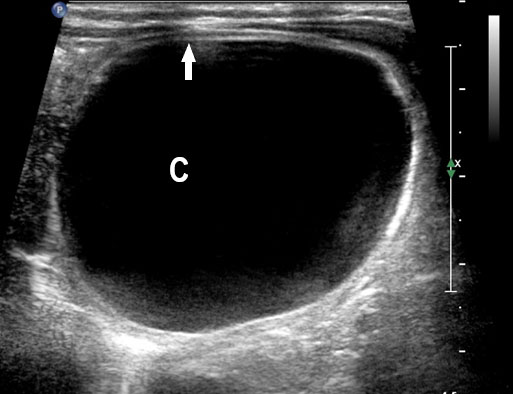
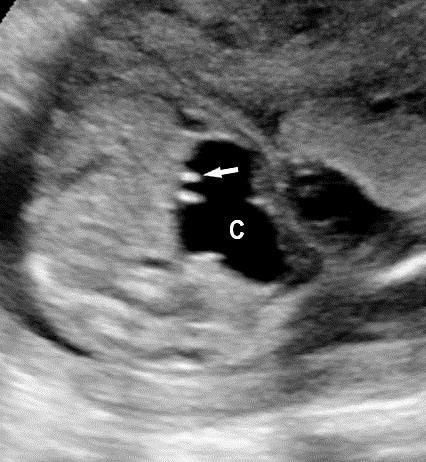
Gastric duplication cysts form 7% of all gastrointestinal duplication cysts. The majority are non-communicating round cysts seen more commonly along the greater curvature of the stomach. They are attached to some part of the stomach and share a common muscle and blood supply. In axial scan of the fetal upper abdomen, sonography shows a thick-walled cyst close to the stomach bubble. It usually shows the two layers of the wall (bowel signature), the mucosal layer being echogenic. Gastric duplication cysts may be seen as a round cystic lesion (Figure 27) or as oval cyst similar in shape to stomach bubble (Figure 28). Postnatally most are asymptomatic. Since they can contain ectopic gastric glands, there can be ulcers that can penetrate and perforate as complications.
(A) |
(B) |
(C) |
(D) |
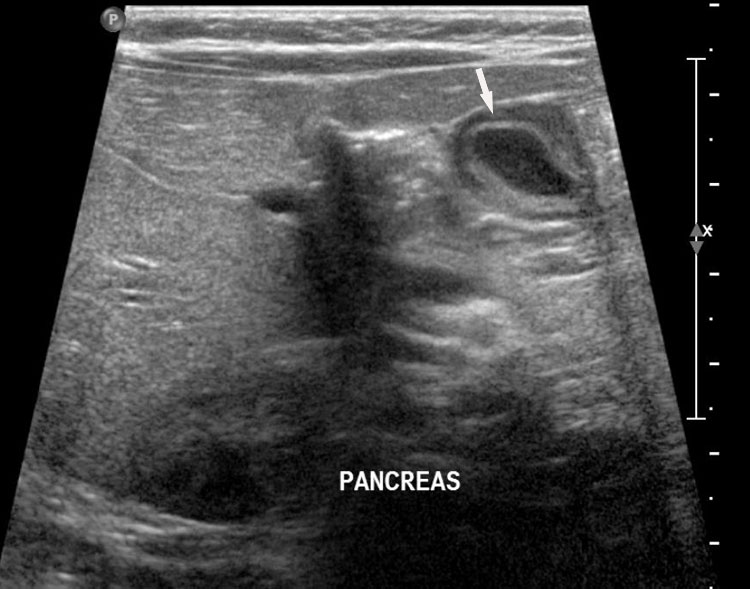
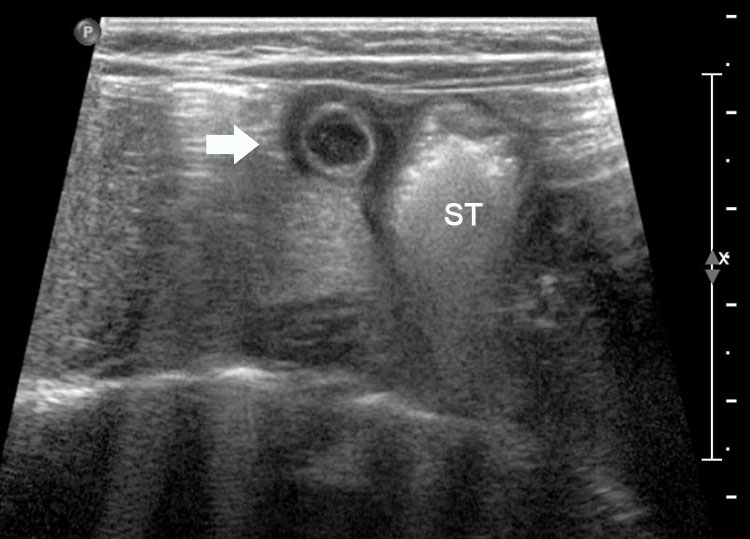
27
Gastric duplication cyst. (A) Axial and (B) coronal scan of fetal abdomen showing a cyst (D) indenting on the distal body of the stomach (ST) with two layer wall (arrow) suggestive of duplication cyst of stomach. (C) Axial and (D) oblique postnatal scans of the newborn showing the typical bowel signature in the wall of the cyst (arrow) which is shared with the wall of body of stomach characteristic of gastric duplication.
(A) |
(B) |
(C) |
(D) |
(E) | |
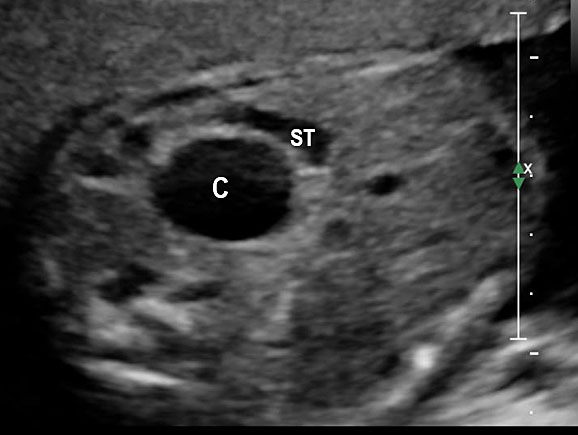
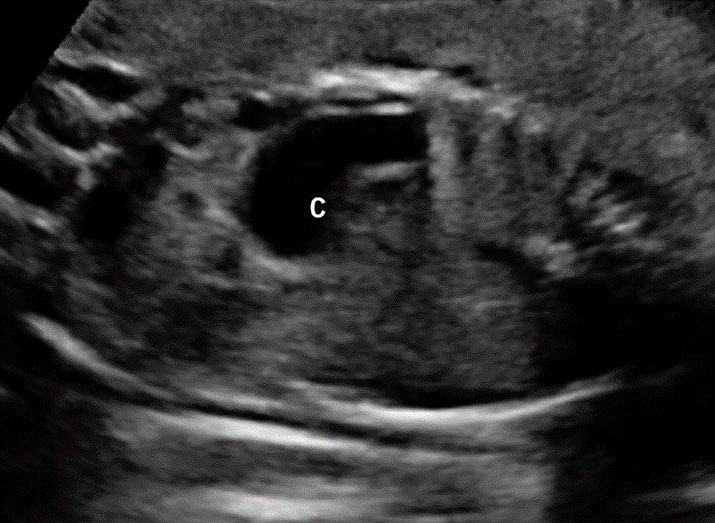
28
Gastric duplication cyst. (A) Axial scan of fetal upper abdomen shows the cyst (C) posteromedial to the stomach bubble (ST). (B) Coronal scan showing the cyst to be of shape of stomach. (C) Oblique scan shows the cyst with gut signature in the wall (arrow). Postnatal sagittal scan (D) confirming the duplication along the greater curvature of stomach with a common wall with it and (E) high-frequency scan depicting the gut signature in the wall of the duplication (arrow).
Small bowel duplication cysts
Small bowel duplications cysts are the most common type of gastrointestinal tract duplication. They can be cystic or tubular (Figure 29). The tubular type may be short or long and communicating with native lumen or non-communicating. They are on the mesenteric side of the bowel in the mesentery. They have the echogenic mucosal lining and share muscle and blood supply with native small bowel. They are seen on the mesenteric side. They may cause intussusception or volvulus in the fetus.
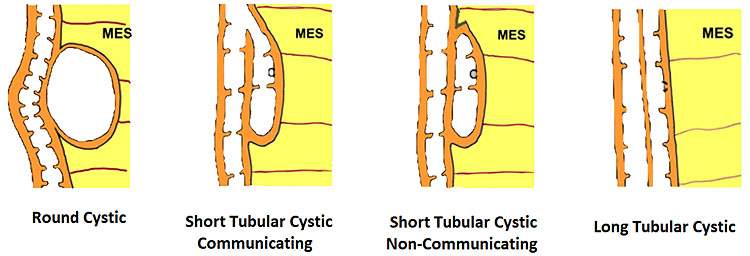
29
Schematic diagram depicting the types of bowel duplications.
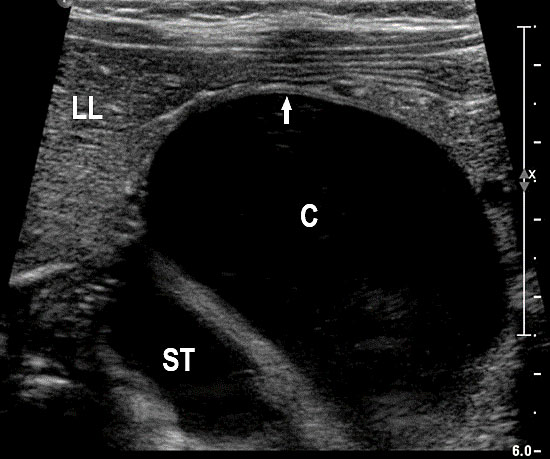
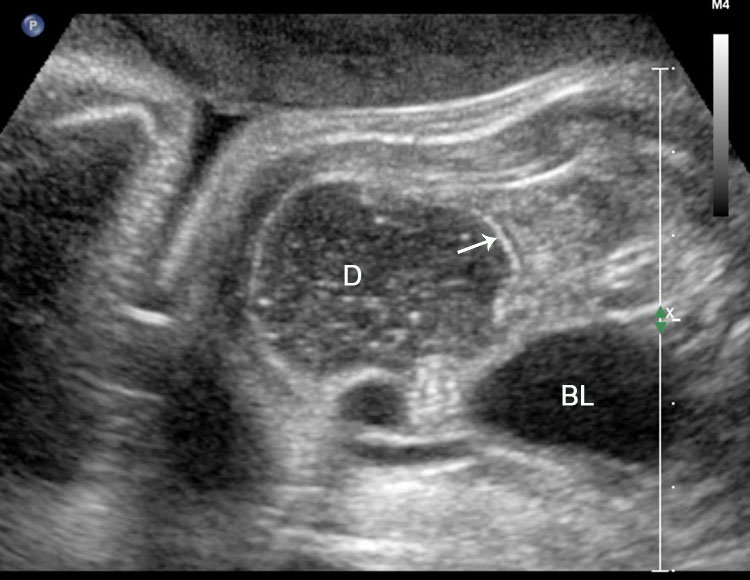
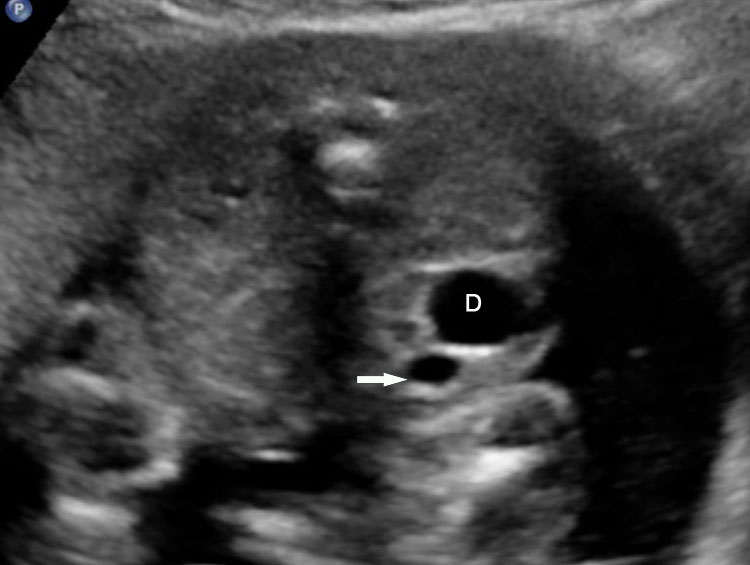
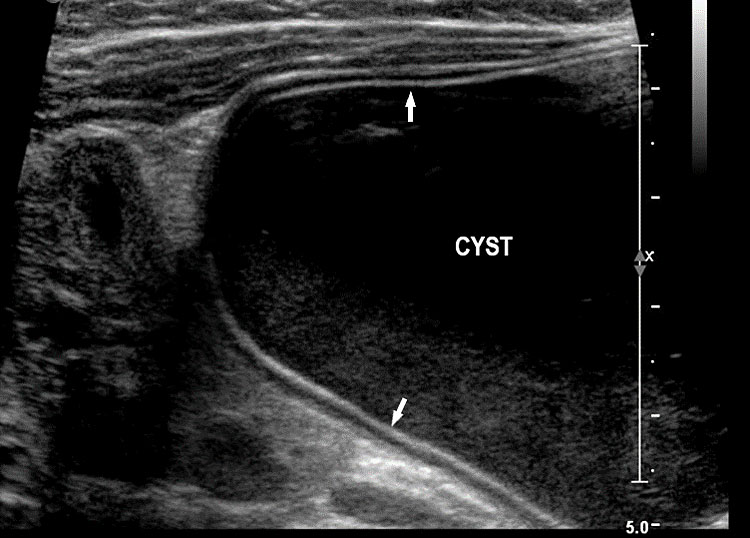
On sonography, the common presentation is an intraabdominal cystic mass (Figure 30A). The signs of origin from known abdominal organs, like kidney, are absent. The appearance of duodenal duplication cysts is similar to that of small bowel duplication except for the location close duodenum and pancreas (Figure 31). The characteristic feature is the two layer sign or the bowel signature in the wall. There is an inner echogenic layer of mucosa and an outer echo-poor muscle in duodenal duplication cysts (Figure 31C) and small bowel duplication cysts (Figure 30B). The small bowel duplication cysts usually show peristalsis which is diagnostic (Video 7). This differentiates duplication cysts from mesenteric cysts and ovarian cysts which do not show peristalsis. They may be seen in changing locations within the abdomen at different periods of scan since they are in the mobile mesentery. The tubular type are seen as a segmental dilated bowel loop with peristalsis (Figure 32 and Video 8). The wall will show the bowel signature. This type has to be differentiated from a dilated loop of bowel in evolving small bowel obstruction. Absence of dilated loops in rest of abdomen and lack of continuity differentiates this from small bowel obstruction. In case of difficulty, a follow-up scan after 2–4 weeks will help, since the tubular duplication will retain the same appearance, whereas small bowel obstruction will show extended dilated loops.
(A) |
(B) |
(C) | |
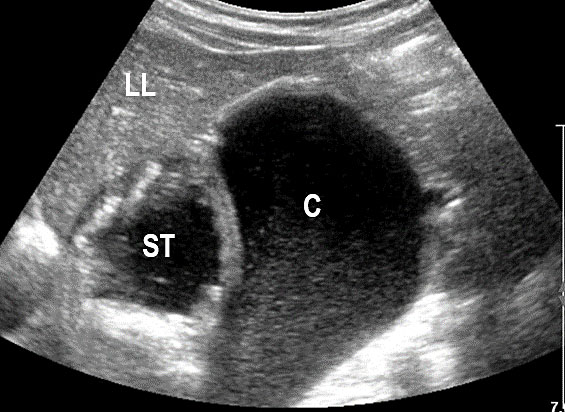
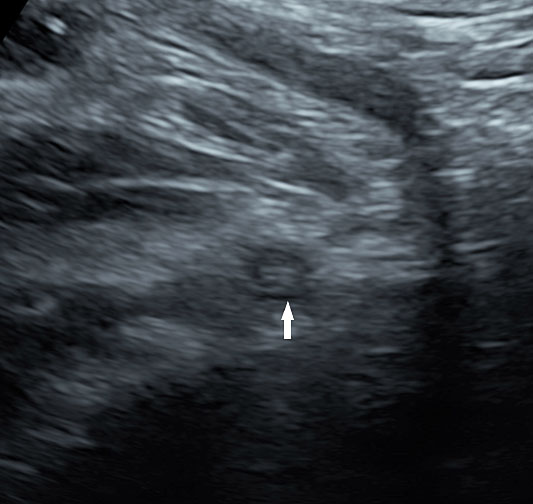
30
Cystic duplication of small bowel. (A) Oblique scan of fetal lower abdomen shows a thick walled spherical cyst (D) anterior and cephalic to urinary bladder (BL). (B) High frequency scan of the cyst (D) shows the bowel signature in the wall and Video 7 shows peristalsis in the cyst, confirming a duplication cyst of small bowel. (C) Postnatal scan confirming cyst (C) with gut signature in the wall.
(A) |
(B) |
(C) | |
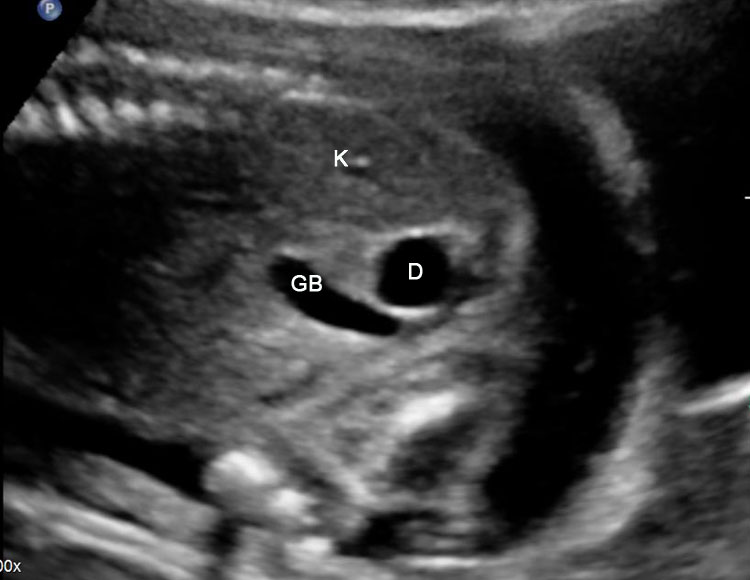
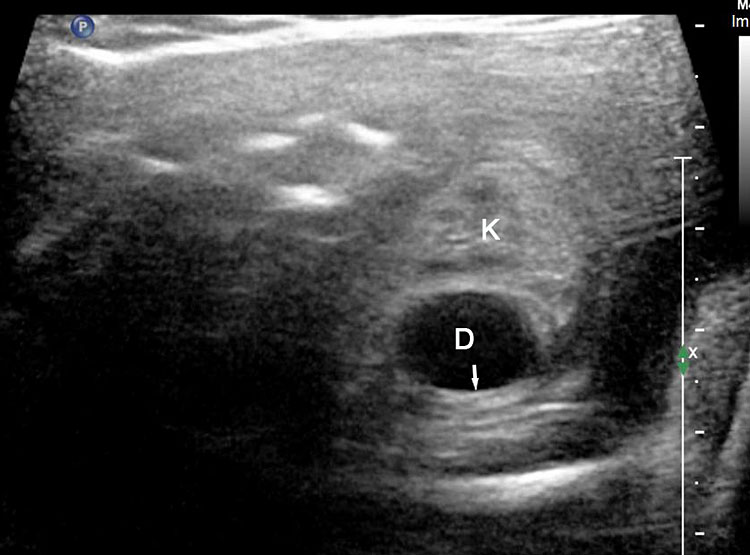
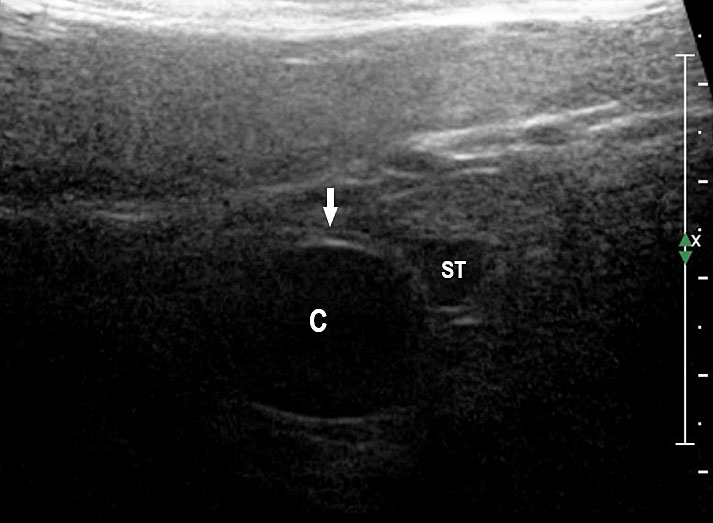
31
Duodenal duplication. (A) Axial and (B) longitudinal scan of fetal upper abdomen showing a cyst (D) between gall bladder (arrow) and the right kidney (K). (C) High frequency scan reveals the bowel signature (arrow) in the wall confirming duplication cyst of duodenum because of location.
7
Peristalsis in the cystic duplication of small bowel.
(A) |
(B) |
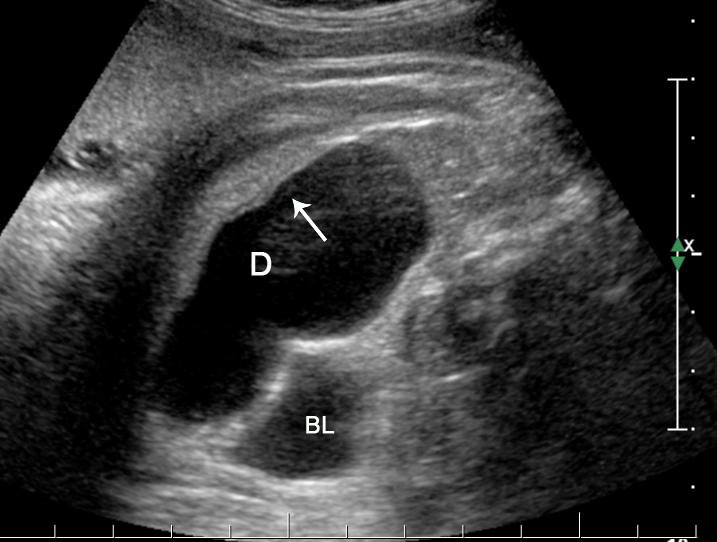
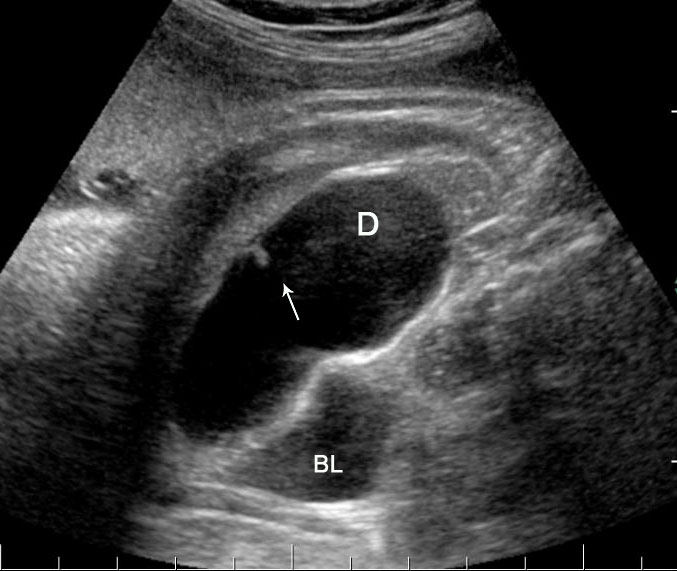
32
Tubular duplication of the small bowel. (A) Axial scan of the fetal lower abdomen shows a focally dilated loop of small bowel (D) with bowel signature in the wall (arrow) anterolateral to urinary bladder (BL). It reveals constriction (arrow) of peristalsis in (B) and peristalsis in Video 8.
8
Tubular duplication of small bowel showing peristalsis.
Large bowel duplication cysts
Large bowel duplication cysts are the rarest form. Sonographic features of large bowel duplication cyst are similar to the small bowel duplications cysts (Figure 33). The only feature that can suggest large bowel duplication cysts, is location of the cyst in the periphery of abdomen. The rarest form of bowel duplication is rectal duplication cyst which presents as a cyst in the pelvis (Figure 34). The location of the cyst between urinary bladder and rectum and the bowel signature in the wall point to this diagnosis.
(A) |
(B) |
(C) |
(D) |
33
Duplication cyst of the large bowel. (A) Axial and (B) coronal scan of the fetal abdomen shows an elongated tubular cystic lesion mimicking a focally dilated loop of bowel (C) in right flank anterolateral to urinary bladder (BL). It reveals constrictions (arrow) of peristalsis in (C) and peristalsis in Video 9. (D) Postnatal scan of right flank shows the bowel signature (arrow) in the wall of the cyst which was a colonic duplication at surgery.
(A) |
(B) |
(C) |
(D) |
(E) | |
34
Duplication cyst of rectum. (A) Axial scan of fetal pelvis shows a cyst (C) posterior and to the left of urinary bladder (BL). (B) Axial scan of lower abdomen shows that the cyst is posterior and to the right. (C) Coronal scan of lower abdomen and pelvis showing the tubular cyst in midline in pelvis and to right in lower abdomen mimicking a dilated rectum. (D) Scan of perineum delineating normal fetal anus. (E) Sagittal scan of fetal pelvis shows the cyst between urinary bladder (BL) and rectum (R) with bowel signature in the wall (arrows).
Small bowel
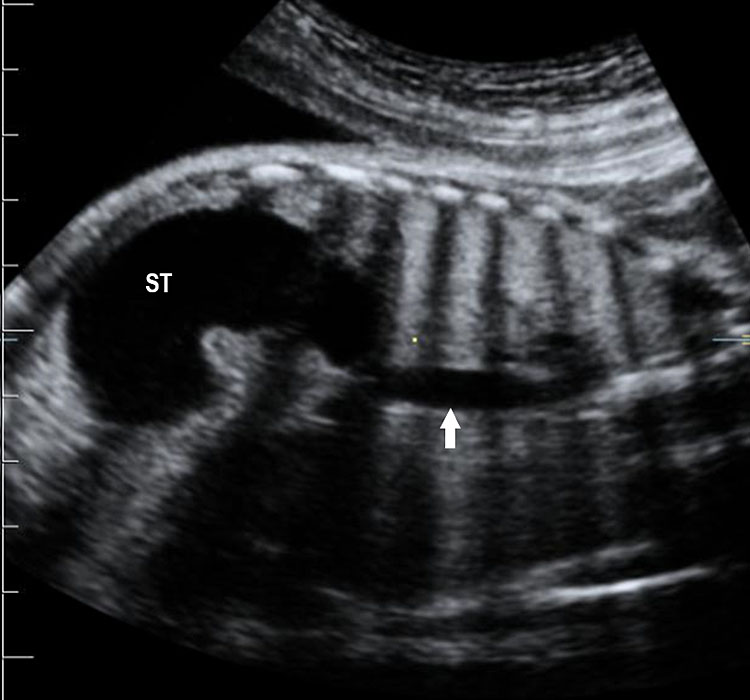
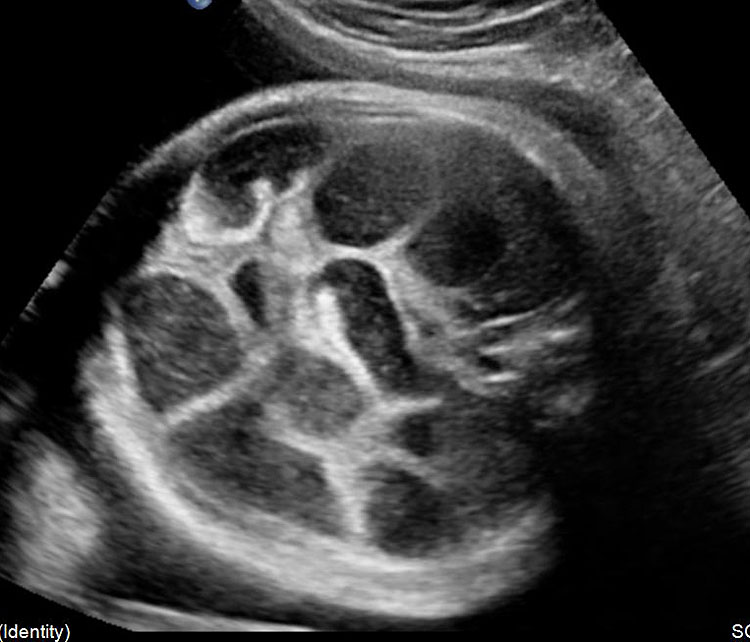
Fetal small bowel loops are seen in the axial section of central abdomen. They are slightly echogenic and fill the central abdomen (Figure 35). With the high resolution of recent machines by transabdominal, small bowel is seen well. The individual serpiginous loops can be made out on high frequency transabdominal scans or endovaginal scan (Figure 35C,D). Occasionally fluid-filled individual segments can be seen in the normal fetus, not exceeding 7 mm in diameter and 15 mm in length (Figure 35E). Active peristalsis of normal small bowel can be observed in later gestation.
(A) |
(B) |
(C) |
(D) |
(E) | |
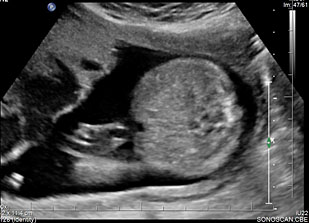
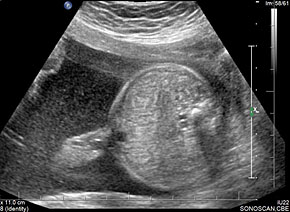
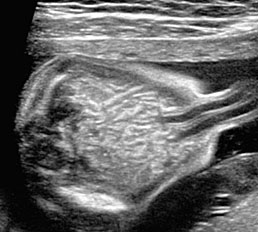
35
Normal fetal small bowel with different transducer frequency. (A) Conventional 3.5 MHz probe showing echogenic bowel filling the mid abdomen (B) with 5 MHz probe: small bowel loops are seen as echogenic curved lines. (C) High frequency 5–12 MHz probe and (D) endovaginal scan show the serpiginous echogenic small bowel loops. (E) Normal fetus showing a fluid distended small bowel of less than 7 mm in diameter.
Small bowel atresia
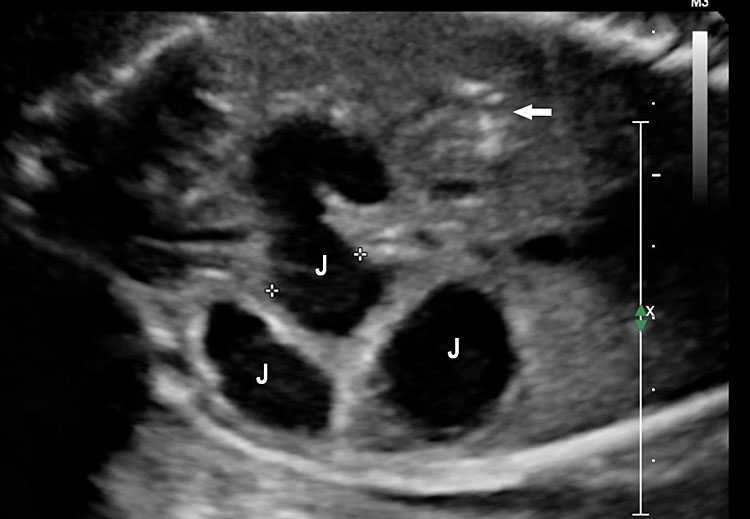
Small bowel atresia (SBA) is a congenital obstruction of the lumen of the duodenum, jejunum or ileum and is one of the most common causes of neonatal bowel obstruction, with an incidence ranging from 1.3 to 2.8 out of 10,000 live births. The pathophysiology of SBA has not yet been completely elucidated. The occurrence of an in-utero vascular accident due to compression, twisting or embolic occlusion of the branches of the mesenteric vessels represents the most likely explanation for this condition. The mortality rate associated with these conditions is usually low, especially in isolated cases; however, neonates with SBA are still affected by significant morbidities such as sepsis, short bowel syndrome and a need for prolonged total parenteral nutrition and hospital stay. The accuracy of detecting jejunal and ileal atresia by prenatal ultrasound has been reported to be variable, and different sonographic criteria for diagnosis, such as polyhydramnios and dilated bowel and stomach, have been described recently. Previously published series have suggested that the detection rate by prenatal ultrasound in cases of non-duodenal small bowel atresia (ND-SBA) varies between 25% and 90%.20 It results in obstruction with proximal dilatation. Incidence is 1 in 3000–8000 live births.
Sonographic features of small bowel obstruction are dilated interconnecting bowel loops with peristalsis or hyperperistalsis (Figure 36 and Video 9). Polyhydramnios is usually present. Small bowel obstruction usually manifests in late second or third trimester.16,17 Dilated stomach can be seen in jejunal atresia. In jejunal atresia there are only a few dilated loops in the upper abdomen (Figure 37). In ileal atresia the whole abdomen is filled with dilated loops, dilatation increasing towards lower abdomen (Figure 38). Small bowel atresia may be secondary to mesenteric volvulus in malrotation of midgut. Here the inversion of superior mesenteric artery and vein can be seen on color Doppler study. In volvulus the real-time “whirl pool” sign can be seen on gray scale and color Doppler study (Figure 39, Videos 10 and 11). The small bowel volvulus may be intermittent without any consequence and may recur in the postnatal period leading to a diagnosis at the time. It can result ischemia and “apple peel” atresia of small bowel (Figure 40 and Video 12). Small bowel obstruction has to be differentiated from congenital chloride diarrhea, which is described subsequently and mesenteric lymphangioma. The multiseptated cyst of lymphangioma can mimic dilated bowel loops but will lack peristalsis. A loop of bowel may be seen to course through the cyst with peristalsis which confirms the diagnosis of lymphangioma (Figure 41 and Video 13). Grossly dilated ureter can mimic small bowel obstruction but can be differentiated by tracing the same to dilated calyces in the kidney (Figure 42). Small bowel atresia may result as a consequence of resolving meconium peritonitis which will be seen as peritoneal calcifications (Figure 43). Multiple atresias of small bowel may follow resolved meconium peritonitis associated with meconium ileus (Figure 44 and Video 14) that may form part of cystic fibrosis.
(A) |
(B) |
36
Small bowel obstruction. (A) Axial scan of mid-abdomen shows multiple dilated small bowel loops with hyperperistalsis seen in Video 10. (B) Oblique scan shows the intercommunicating dilated small bowel loops.
9
Small bowel obstruction: dilated small bowel loops filling the fetal abdomen with hyperperistalsis.
(A) |
(B) |
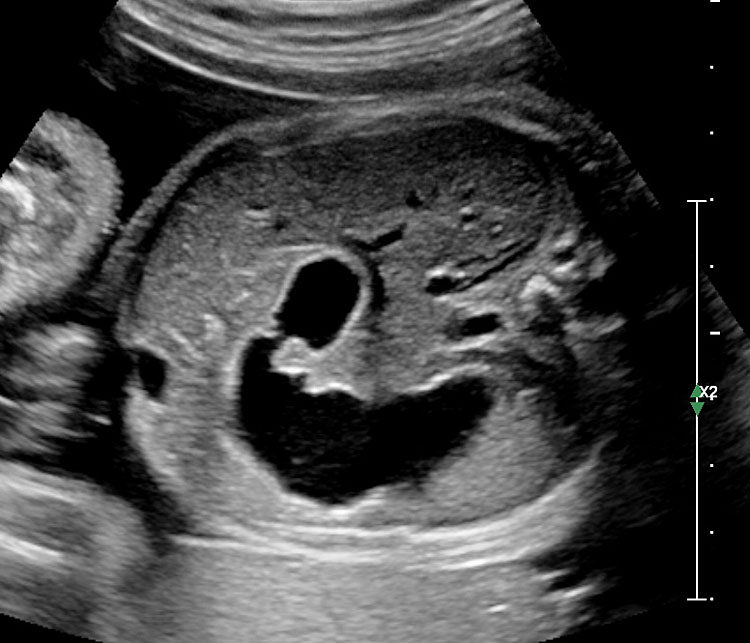
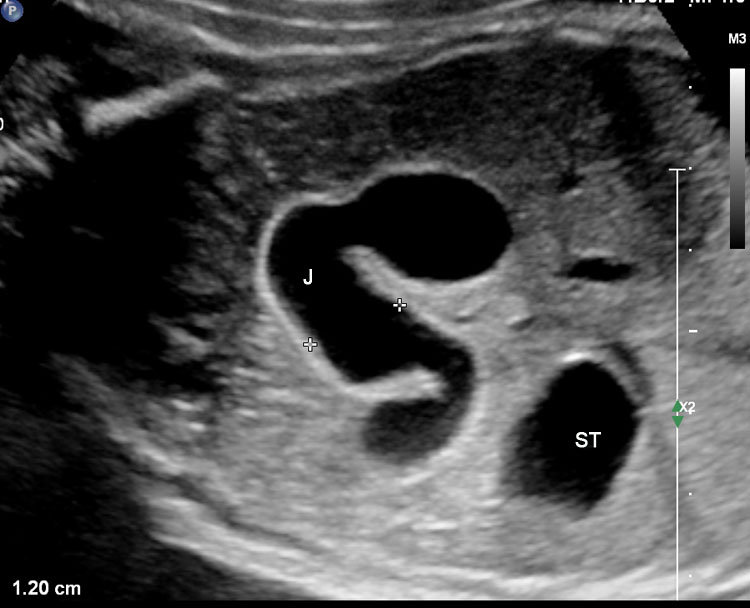
37
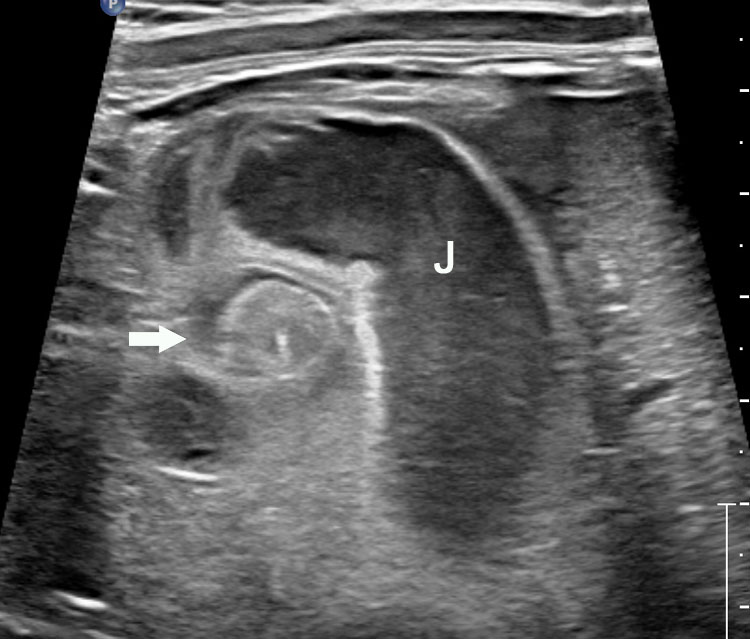
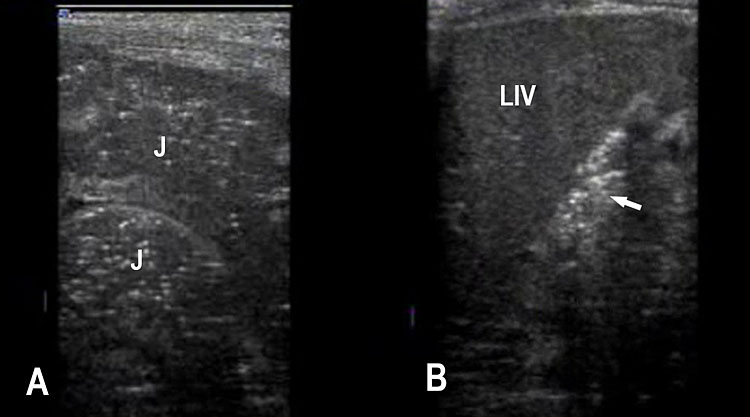
Jejunal atresia. (A) Axial scan of upper abdomen shows dilated stomach and duodenum. (B) Coronal scan shows a few dilated jejunal loops (J).
(A) |
(B) |
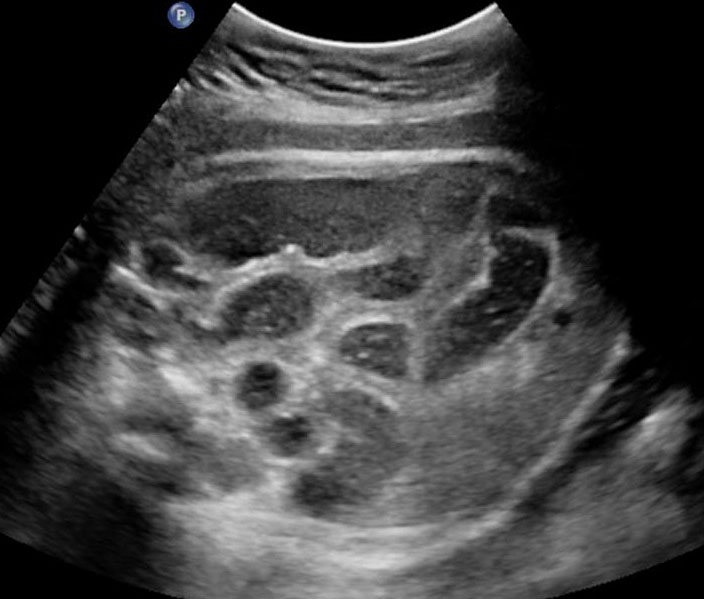
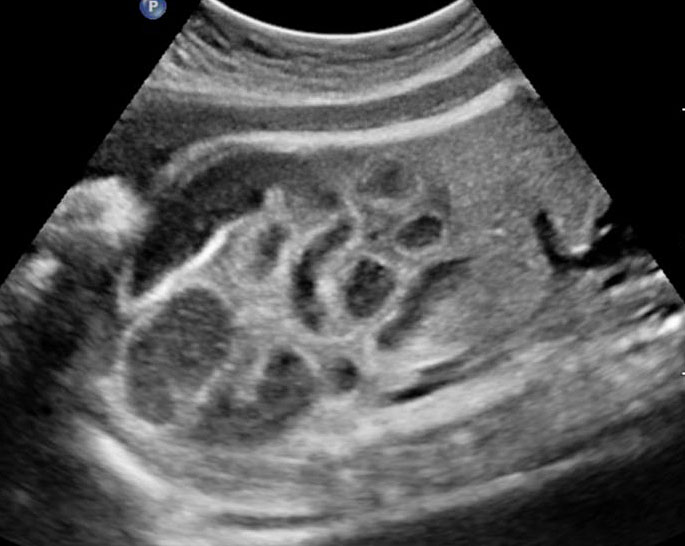
38
Ileal atresia. (A) Axial scan shows multiple dilated small bowel loops filling the abdomen. (B) Longitudinal scan shows increasing dilatation towards lower abdomen indicating ileal atresia.
(A) |
(B) |
(C) | |
39
Small bowel atresia due to malrotation of midgut with volvulus. (A) Axial scan showing the dilated small bowel loops with a coffee bean appearance. (B) High frequency oblique scan just below the dilated loop shows the mass of “whirl pool” in gray scale and (C) color Doppler. Videos 11 and 12 show the “whirl pool” sign when the probe is moved down confirming the volvulus on gray scale and color Doppler study.
10
Movement of transducer showing the “whirl pool” sign of malrotation of midgut with volvulus in grayscale (Figure 14) and color Doppler study (Figure 15).
11
Movement of transducer showing the “whirl pool” sign of malrotation of midgut with volvulus in grayscale (Figure 14) and color Doppler study (Figure 15).
(A) |
(B) |
(C) | |
40
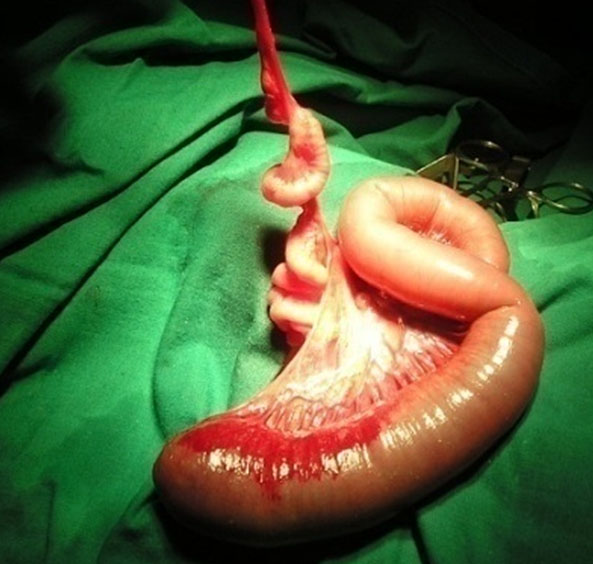
Apple peel small bowel atresia due to volvulus. (A) Oblique scan of fetal upper abdomen shows multiple dilated jejunal loops. (B) Axial scan shows the “whirlpool” on color Doppler by the side of dilated bowel. (C) Surgical photo shows the apple peel atresia (Video 13).
12
Apple peel atresia due to volvulus showing the “whirl pool” sign.
(A) |
(B) |
(C) | |
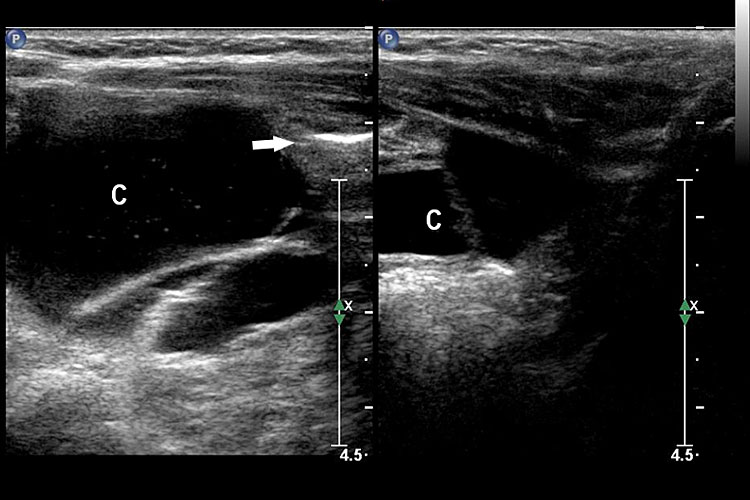
41
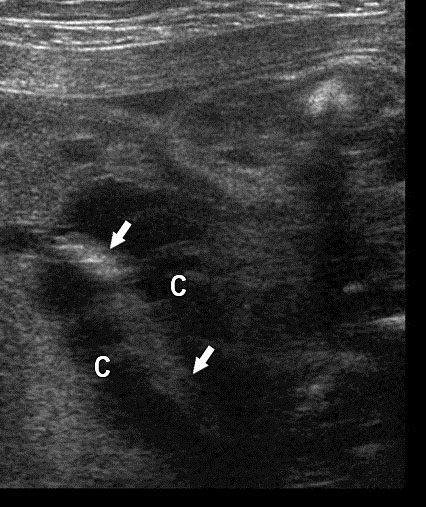
Mesenteric lymphangioma. (A) Axial scan of fetal midabdomen showing the multiseptated cyst (arrows) mimicking dilated bowel loops. (B) High frequency scan shows a small bowel loop (arrows) coursing through the cyst (C) (Video 14). (C) Postnatal scan confirming the mesenteric lymphangioma (C) with the loop of bowel (arrow) in it.
13
Mesenteric lymphangioma shows peristalsis in bowel loop coursing through the cyst.
(A) |
(B) |
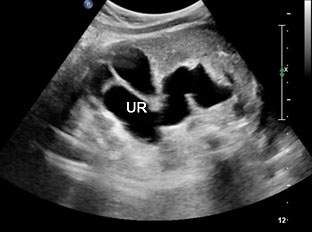
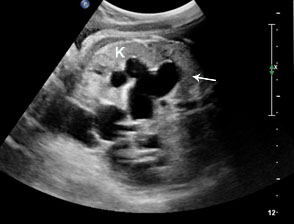
42
(A) Oblique axial scan of fetal abdomen shows grossly dilated ureter mimicking dilated small bowel but on tracing, it is connected to the dilated renal pelvis and calyces in the kidney confirming that it is grossly dilated ureter.
(A) |
(B) |
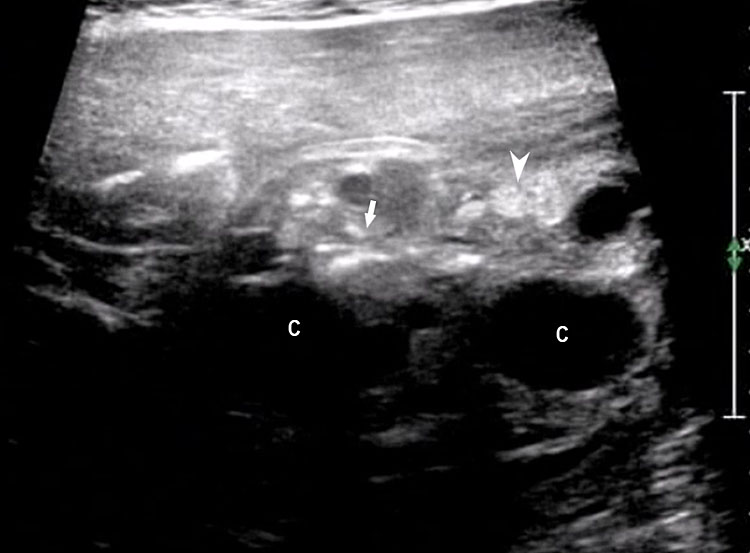
43
(A) Coronal scan of fetal abdomen shows a few loops of dilated jejunum (J) and peritoneal calcifications (arrow). Postnatal scan confirms jejunal atresia (A) with peritoneal calcifications (B) of resolved meconium peritonitis.
(A) |
(B) |
(C) | |
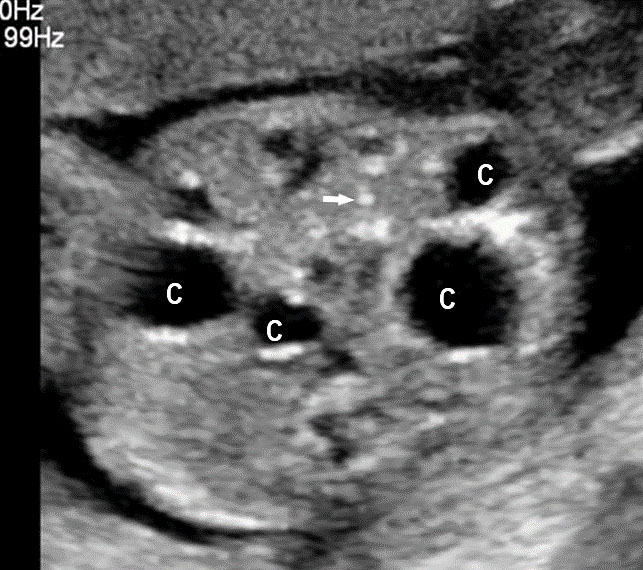
44
Multiple small bowel atresia. (A) Axial scan of fetal midabdomen showing multiple cystic lesions (C) with peritoneal calcifications (arrow). (B) High frequency scan shows multiple cysts (C), peritoneal calcifications (arrow) and echogenic bowel (arrowhead) confirmed by peristalsis in Video 15.
14
Peristalsis in the echogenic bowel in meconium ileus.
Congenital chloride diarrhea
Congenital chloride diarrhea (CCD) is a very rare disorder, reported mainly in Finland. It is an autosomal recessive disorder caused by mutations in the SLC26A3 gene on chromosome. The underlying defect is in chloride resorption by intestine associated with impaired transport of bicarbonate. This results in hygroscopic retention of fluid in the intestine leading to watery diarrhea and polyhydramnios.21
Sonography reveals distension of the fetal abdomen with uniformly dilated small and large bowel filling the abdomen with hyperperistalsis (Figure 45 and Video 15). There is polyhydramnios due to fetal diarrhea. Differential diagnosis is ileal atresia and anal atresia. There is uniform dilatation of bowel in CCD, while the dilatation increases distally in ileal atresia. The fluid distended rectum can be seen in CCD, while it is not seen in ileal atresia and occasionally the fetal diarrhea can also be seen as fluid gushing out from fetal anus (Video 16). Normal fetal anus will be seen, differentiating it from anal atresia.
(A) |
(B) |
(C) | |
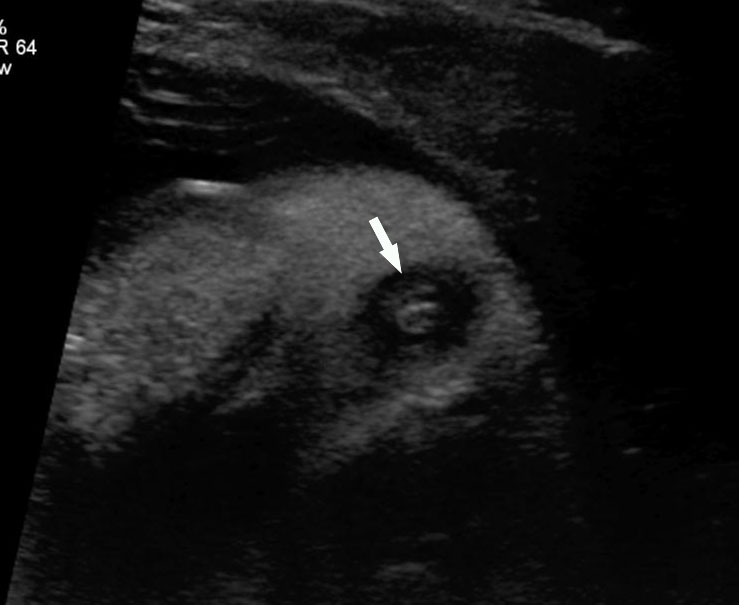
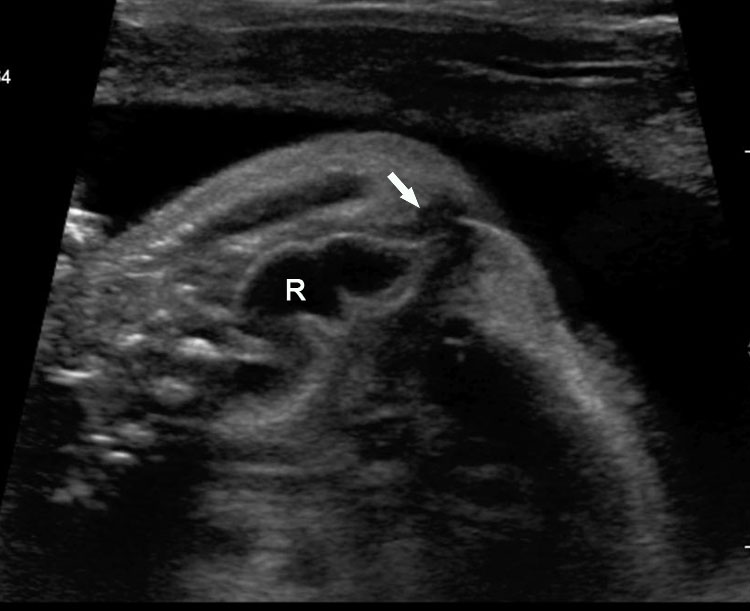
45
Congenital chloride diarrhea. (A) Axial scan of the fetal abdomen shows distension of abdomen with uniformly dilated bowel loops with polyhydramnios. They show intense peristalsis as shown in Video 16. (B) Tangential scan of perineum shows the fetal anus ruling out anal atresia. (C) Sagittal scan of fetal pelvis and perineum showing the fluid distended rectum (R) that differentiates it from ileal atresia and the anal canal (arrow) reaching up to the skin of perineum. Video 17 shows the fetal diarrhea as gush of fluid from the anus.
15
Congenital chloride diarrhea. Axial scan of fetal abdomen showing uniformly dilated bowels distending the fetal abdomen with intense peristalsis.
16
Congenital chloride diarrhea. Sagittal scan of fetal perineum showing fetal diarrhea as a gush of fluid from fetal anus.
Echogenic bowel
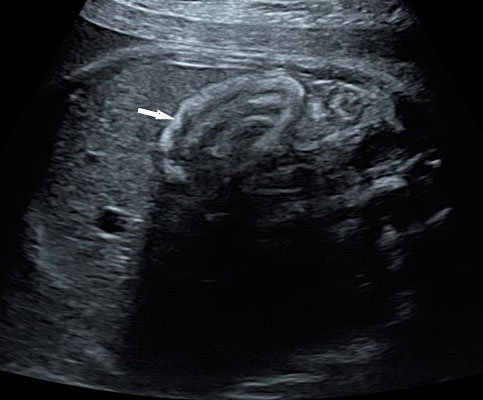
On fetal sonography echogenic bowel (EB) is diagnosed when bowel appears echogenic in different scan planes, across various transducer frequencies without harmonics and remains echogenic equal to bone when the gain is decreased to see only bones (Figure 46, Videos 17 and 18). It is detectable at the time of the second trimester routine antenatal ultrasound in 0.2–1.8% of all fetuses.22 Echogenic bowel can be a normal variant or may be due to normal echogenic meconium near term (Figure 49). Abnormal situations of echogenic bowel are swallowed blood, a marker for trisomy 21, feature of cystic fibrosis, cytomegalovirus infection and fetal growth restriction. In fetuses with EB, the sonographic survey must be complete to exclude associated sonographic soft markers for trisomy 21, bowel dilation, fetal ascites, and peritoneal calcification or extra-intestinal anomalies that may correlate with an increased risk of adverse neonatal outcome.
(A) |
(B) |
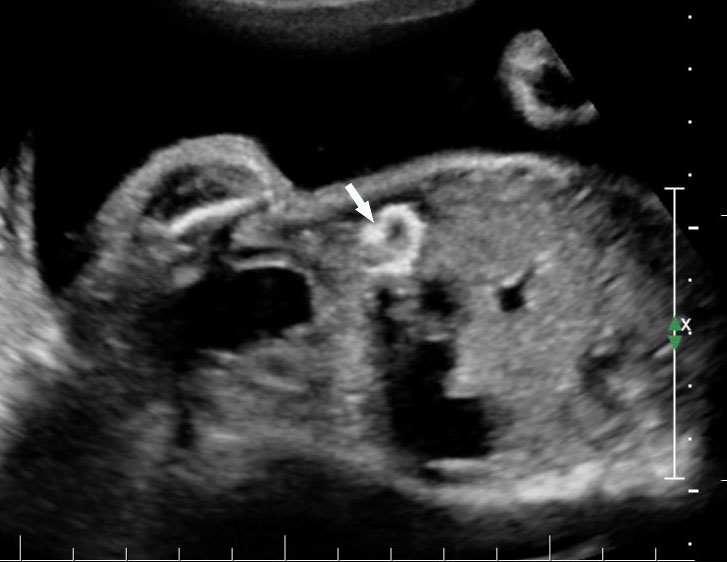
46
Coronal scan of fetal abdomen showing echogenic bowel (A) which remains echogenic equal to bone with decrease in the gain (B) (Videos 18 and 19).
17
Coronal scan of fetal abdomen with gradual decrease of gain showing echogenic bowel disappearing in normal (Figure 21) and persisting equal to bone in echogenic bowel (Figure 22).
18
Coronal scan of fetal abdomen with gradual decrease of gain showing echogenic bowel disappearing in normal (Figure 21) and persisting equal to bone in echogenic bowel (Figure 22).
Cystic fibrosis
Cystic fibrosis (CF) is a life-threatening monogenic recessive disorder caused by biallelic pathogenic variants in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7. CF is characterized by chronic bacterial airway and sinus infection, pancreatic exocrine insufficiency leading to incomplete breakdown of fats and meconium ileus, congenital bilateral absence of the vas deferens (CBAVD) in males (leading to infertility) and a high sweat chloride concentration. Progressive respiratory disease is the major cause of mortality and morbidity among young CF patients. Normally, the CFTR protein pumps chloride ions out into the thick mucus of the digestive and respiratory tracts, which helps pull water into the mucus to make it less viscous. When the CFTR protein is not working properly, chloride becomes trapped inside the cell, preventing water from thinning out the body’s secretions. As a result, the mucus secreted by these cells is unusually thick and sticky. This mucus builds up and obstructs the organs where it is secreted. The inspissated meconium obstructs the small intestine at the level of the terminal ileum – meconium ileus. The meconium-distended segments of ileum can give way to complications like prenatal volvulus, ischemic necrosis, intestinal atresia, or perforation and extrusion of the meconium into the peritoneum causing meconium peritonitis. Loss of CFTR function may contribute to pH dysregulation resulting in injury of the biliary tree. The incidence of CF is greater than 1 in 2000 births in Scotland and Ireland, is approximately 1 in 3500 births in the United States, and is only 1 in 40,000–100,000 births in India. Almost 2000 different CFTR variants have been identified, though 85% of the disease burden is attributable to only a few variants. The most common pathogenic variant in the CFTR gene is a three–base pair deletion at position 508 resulting in the loss of phenylalanine (F508del). This variant accounts for about 70% of pathogenic CFTR variants globally. Prenatal diagnosis can be offered when both parents are known carriers of CF causing mutations. Even in the absence of a family history of cystic fibrosis, CF can be suspected during prenatal period in the presence of ultrasound digestive abnormalities such as fetal hyperechogenic bowel (FEB) and intestinal loop dilatation, mostly during the second trimester of pregnancy.23,24,25
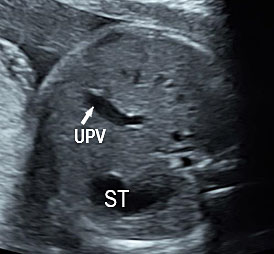
As a first step, a screening for frequent mutations according to geographical origin should be undertaken, and if one parent is positive, an exhaustive study of CFTR should be done in the other parent. There is a high risk of cystic fibrosis in fetuses presenting with the triad associating FEB, intestinal loop dilatation, and non-visualized gall bladder (NVFGB) (Figure 47).25 In order to minimize the risk of CF for couples of geographical origins in which CFTR variance is understudied or poorly understood, performing an extensive study of the CFTR gene is appropriate.23
(A) |
(B) |
(C) | |
47
(A) Axial and (B) coronal scan of fetal upper abdomen showing non-visualized gall bladder. (C) Axial scan of mid-abdomen shows echogenic bowel. Fetus tested homozygous for CFTR gene for cystic fibrosis.
Fetal large bowel
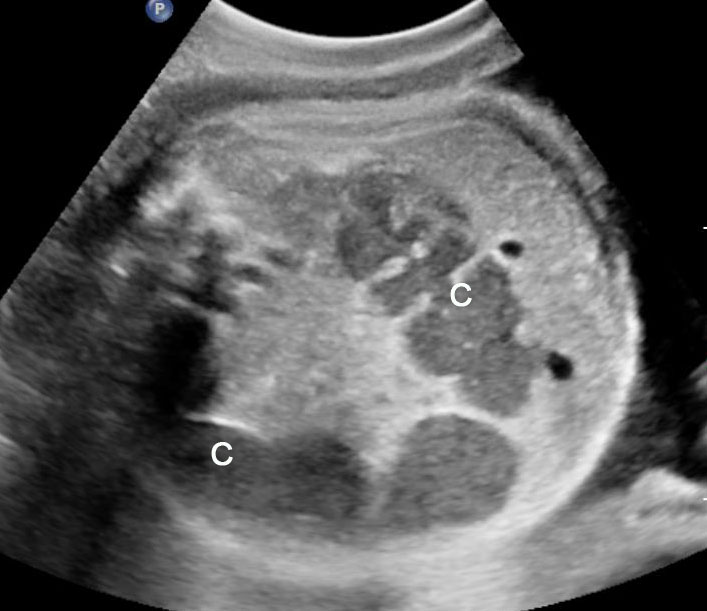
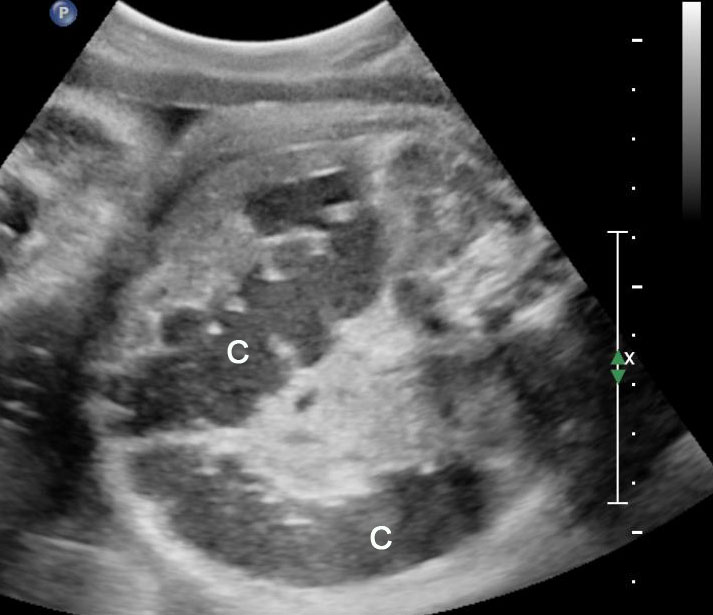
Fetal colon becomes visible on sonography in late second trimester and is clearly seen close to term. It is seen as a hypoechoic tubular structure with haustrations in periphery of fetal abdomen in the usual location of colon. There are redundancies and turns (Figure 48). Occasionally, colon may be filled with echogenic meconium near term in normal fetuses (Figure 49). The diameter of colon shows a large range – up to 18 mm near term. Dilated colon with echogenic meconium balls is a feature of anal atresia (Figures 54B and 59A).
(A) |
(B) |
48
(A) and (B) Axial scans of fetal abdomen in third trimester showing the normal fluid distended colon (C) along the periphery of abdomen with haustrations and redundancies.
(A) |
(B) |
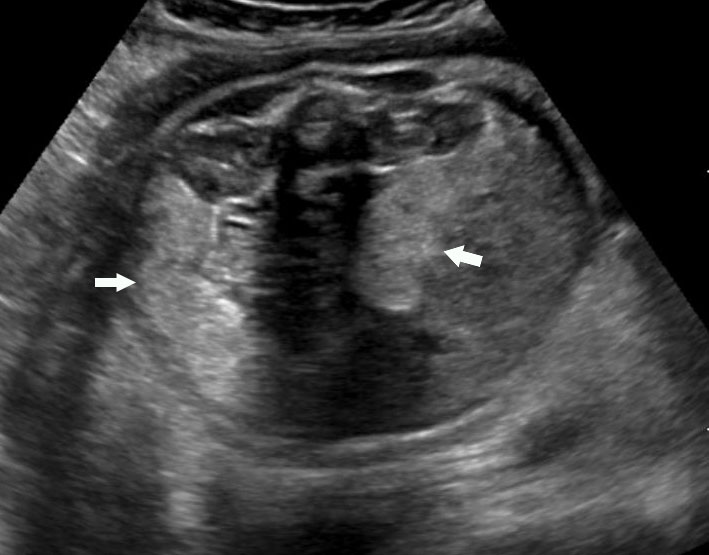
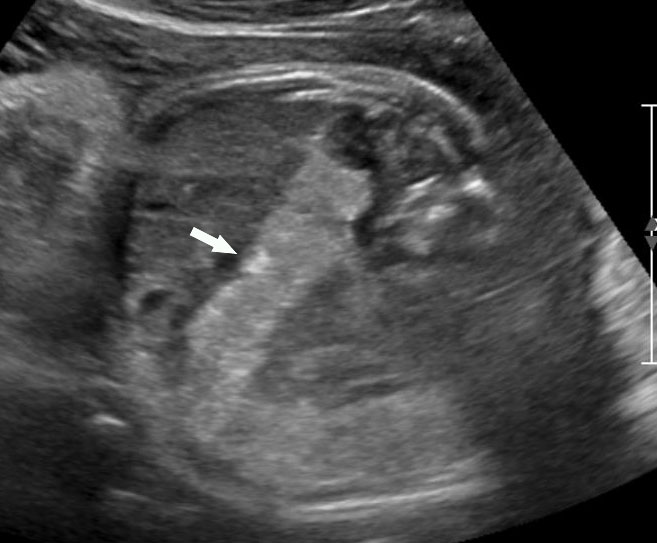
49
(A) and (B) Axial scans of abdomen of 35 weeks fetus shows normal colon (arrow) filled with echogenic meconium.
Anorectal malformation
Anorectal malformation (ARM) is a group of anomalies with absent anal opening. It is a spectrum of anomalies from minor types to very complex anomaly. It may be a low anorectal malformation in the form of covered anus where the anus is covered by skin only. In high anorectal malformation the rectum is high above the perineum, most of the times complicated by a fistula with bladder, urethra or vagina. The anomaly may be more complex in the form of urorectal septal malformation (URSM) sequence. It may also form part of complex associated anomalies like VACTERL association, caudal dysplasia, trisomy 18 and 21, OEIS complex (comprising omphalocele, exstrophy of the cloaca, imperforate anus, and spinal defects) or bladder exstrophy complex. Anorectal malformation is seen in about 1 in 5000 live births.26,27,28 Prenatal recognition of this pathology is important to inform the parents and to improve the management during pregnancy and in neonates.
Sonographic features
Initially diagnosis of ARM is suspected when the fetus shows a dilated colon with or without echogenic meconium balls in it (Figures 54B and 59A). The sensitivity of the sonography in diagnosis of anorectal malformation is very poor ranging from 0 to 33%. The reasons were: (1) the wide range of size of normal large bowel; (2) difficulty in differentiating the small bowel from large bowel in earlier gestation; and (3) the colon is not dilated in many cases of ARM. Hence the diagnosis of ARM is improved to a great extent by direct visualization of the fetal anus. The evaluation of the fetal perineum is not included in most of the guidelines.
Technique of seeing the fetal anus
From the axial scan of pelvis showing the urinary bladder, the axial scan is continued down up to the perineum. The tangential axial scan of perineum is obtained by tilting and rotating the transducer to see the two gluteal regions with the anus in between (Figure 50, Videos 19 and 20). The anus is seen as an echo-poor circle due to the anal muscle complex with an echogenic dot in the center due to the lumen. In this section the ischium should not be seen to confirm that the section is at the correct plane of section of perineum. The nonvisualization of anus by this technique is diagnostic of anorectal malformation (Figure 51 and Video 21). A high frequency probe can be used in doubtful cases to obtain sagittal and coronal scans of fetal pelvis and perineum to see the rectum and anal canal reaching up to the perineal skin (Figure 50D,E).26 Then, the scan is extended to look for other associated anomalies. When an anomaly like a spinal defect is seen, the fetal anus is looked for to diagnose a complex anomaly like VACTERL association.
(A) |
(B) |
(C) |
(D) |
(E) | |
50
Technique of seeing fetal anus. Transducer is moved down from (A) axial section of pelvis through the urinary bladder (BL) and sacral spine to (B) the lower pelvis at the level of ischium and then (C) the tangential section of the perineum showing the anus (arrows) (Videos 20 and 21). (D) Sagittal high resolution scan showing the fetal anus as an echo-poor tubular structure with central echogenic stripe (arrow). R, rectum. (E) Coronal high resolution scan showing the fetal anus (arrows) and rectum (R).
19
Technique of seeing fetal anus in female fetus (16) and male fetus (17).
20
Technique of seeing fetal anus in female fetus (16) and male fetus (17).
(A) |
(B) |
(C) | |
51
Failure to see the fetal anus in a fetus with VACTERL anomaly. (A) Tangential scan of the perineum shows the midline echogenic stripe of the perineum with non-visualized anus (Video 22). (B) Photograph of the perineum of the fetus shows the anal atresia. (C) Coronal scan of the fetal spine showing the hemi vertebra (arrow).
21
Showing the nonvisualized fetal anus.
Nonvisualized fetal anus
The fetal anus is not visualized in anorectal malformation (ARM). The ARM can be isolated or form part of complex associations of which the commonest is URSM sequence.
Urorectal septal malformation (URSM) sequence or cloacal malformation
The lower portion of gastrointestinal and the genito-urinary tracts develop from the caudal mesoderm – the primitive cloaca. Between 5 and 7 weeks of gestation, the urorectal septum develops and descends down dividing the cloaca into the anterior urogenital sinus and posterior hindgut and fuses with the cloacal membrane. The urorectal septum then breaks down and leaves an open urogenital sinus and rectum. The urogenital sinus differentiates into the urinary and genital tracts. Arrest of this developmental process would lead to URSM, though a precise etiology has not been established. No familial inheritance has been reported. Recent evidence suggests the involvement of homeobox and sonic hedgehog signaling pathways in the pathogenesis of URSM. The genetic cause has not yet been established. The incidence of URSM is reported to be 1 in 50,000 to 250,000 among live born with a female preponderance.27,28
The following signs would suggest a URSM:
- Non-visualization of the anus on a tangential scan of the fetal perineum (Figure 51).
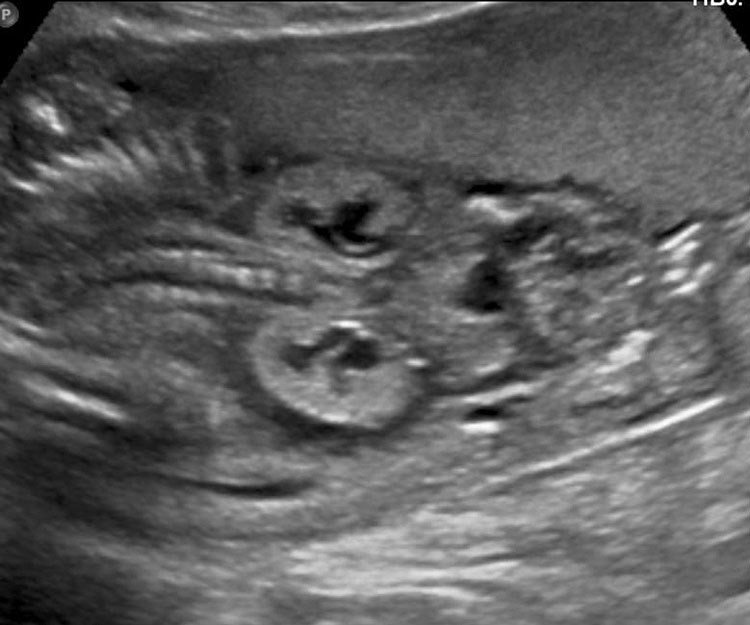
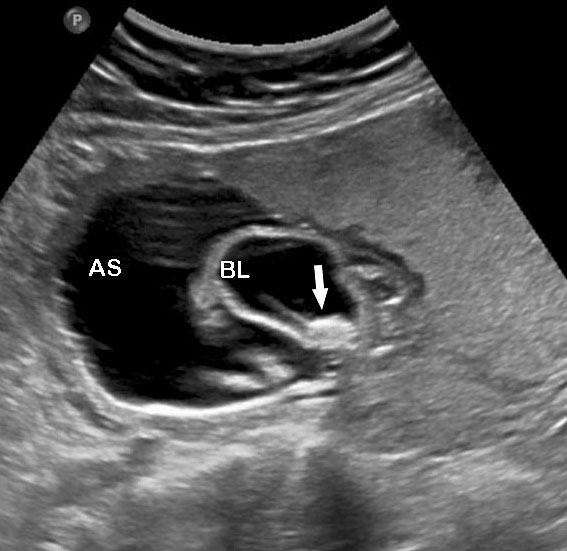
- Cystic mass with a fluid-debris level that funnels to the pelvis in a female fetus representing the fluid distended common urogenital sinus (cloaca) is one of the most consistent observations (Figure 52). The fluid-debris level is due to the urine admixed with meconium distending the urogenital sinus of the fetus. Mullerian duplication is noted in 60% of cases with URSM. The incidence increases to 80% in those cases where prenatal diagnosis of septated cystic mass is made, which represents hydrocolpos of duplicated vagina (Figure 53). Hydrocolpos is highly diagnostic of URSM, but its absence does not rule out URSM. Hydrocolpos may not be evident in up to 50% of fetuses with URSM.
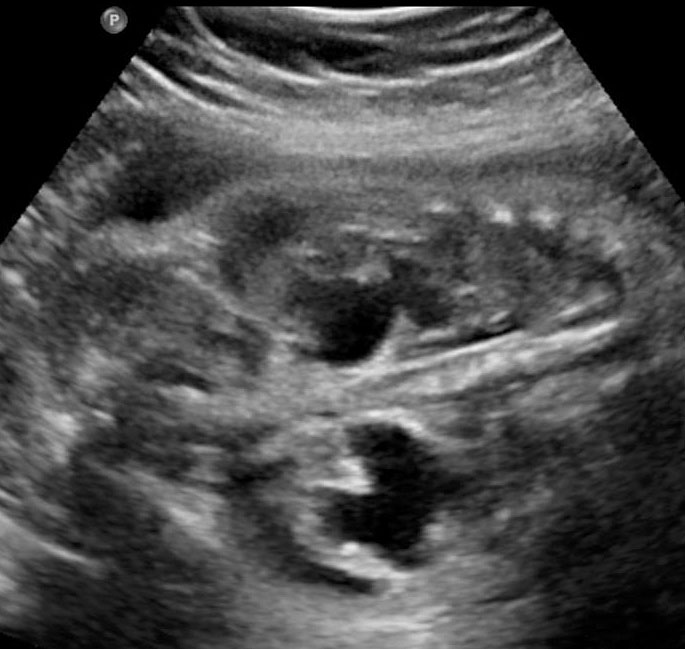
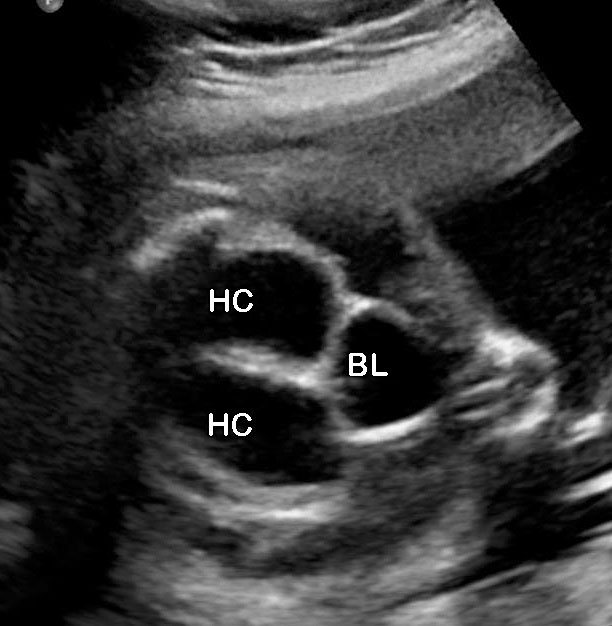
- Renal anomalies are reported in 90% of fetuses with URSM. Bilateral hydronephrosis (Figures 52, 53, 55 and 56) is the most common, though other anomalies such as renal cystic dysplasia (Figure 54), pelvic kidney, horse-shoe kidney and crossed fused ectopia have also been noted.
- There may be dilated colon with echogenic balls of meconium (Figures 54 and 59).
- Echogenic ascites due to the reflux of urine mixed with meconium through the Fallopian tubes into the peritoneum (Figure 53) may be seen. Occasionally meconium peritonitis may be visible as peritoneal calcification. Bowel may be either normal in caliber or dilated.
- Spinal abnormalities such as hemivertebra or hypoplastic sacrum are frequently associated (Figure 51).
- The degree of oligohydramnios depends on the severity and renal involvement. Complete URSM is associated with anhydramnios.
(A) |
(B) |
52
Urorectal septal malformation sequence (URSM): (A) Coronal scan of fetal abdomen reveals anhydramnios and bilateral hydronephrosis with echogenic parenchyma. (B) Demonstrates ascites and pyriform cystic mass (C) with debris (arrow).
(A) |
(B) |
(C) |
(D) |
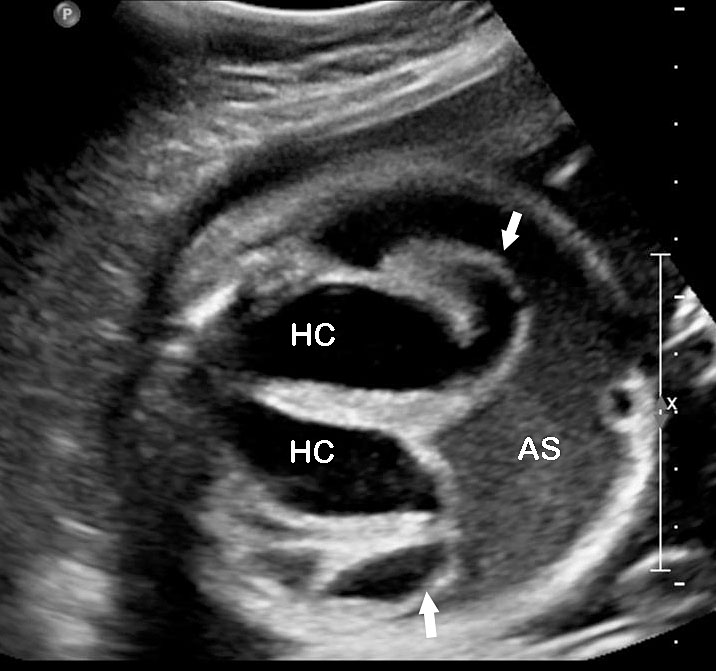
53
Urorectal septal malformation sequence (URSM). (A) Coronal scan shows bilateral hydronephrosis. (B) Axial scan of fetal pelvis shows a large septated cystic mass (HC) posterior to urinary bladder (BL). (C) Oblique coronal scan of fetal abdomen demonstrates echogenic ascites (AS) and hydrocolpos (HC) of duplicated vagina with hydrometra of the duplicated uteri (arrows). (D) Tangential scan of fetal perineum demonstrates nonvisualized anus.
(A) |
(B) |
(C) | |
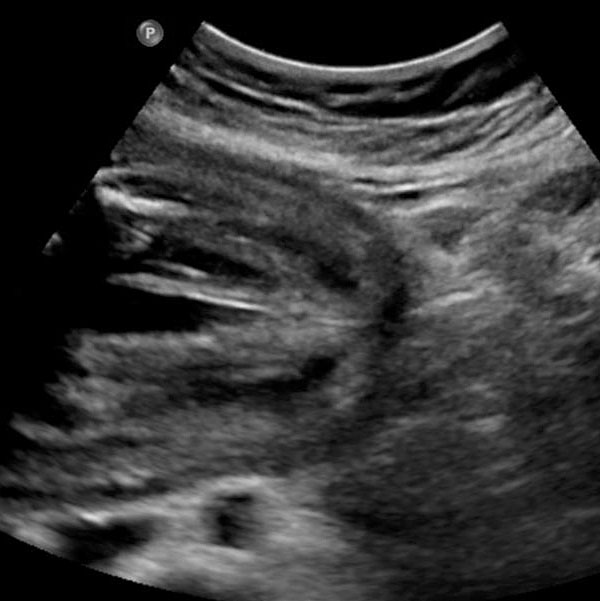
54
Urorectal malformation sequence (URSM). (A) Coronal scan reveals multicystic dysplastic of the kidney. (B) Axial scan of lower abdomen shows dilated colon with echogenic meconium balls. (C) Tangential scan of fetal perineum demonstrates nonvisualized anus.
There could be other associated anomalies such as ambiguous genitalia (Figure 57C) and single umbilical artery. Antenatal recognition is important since it may provide the couple with the option of termination when diagnosis is established before the legal time limits of termination. URSM carries a guarded prognosis. Functional disturbances are often encountered in anorectal, urological and gynecological systems after corrective surgery.
Two types of URSM have been described. Complete URSM is characterized by the absence of anal and perineal openings, a smooth perineum and ambiguous genitalia (Figure 55). It is a lethal anomaly and would result in stillbirth and perinatal mortality. Partial URSM is characterized by a single anal or perineal opening that serves a common conduit for gastrointestinal and genitourinary tracts (Figures 56 and 57). It is a less severe variant.26,27,28 Differentiation of the types is not possible by prenatal ultrasonography. In both types fetal anus will not be seen. The association of URSM with other anomalies such as VACTERL and OEIS complex has been described.
(A) |
(B) |
(C) |
(D) |
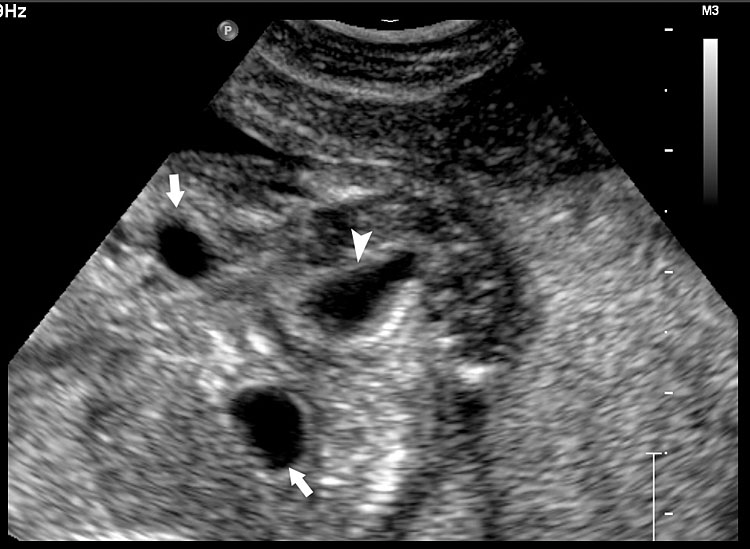
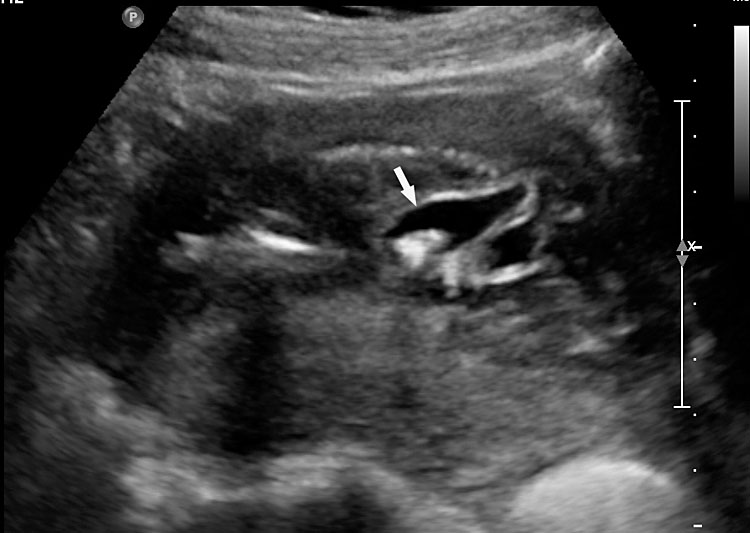
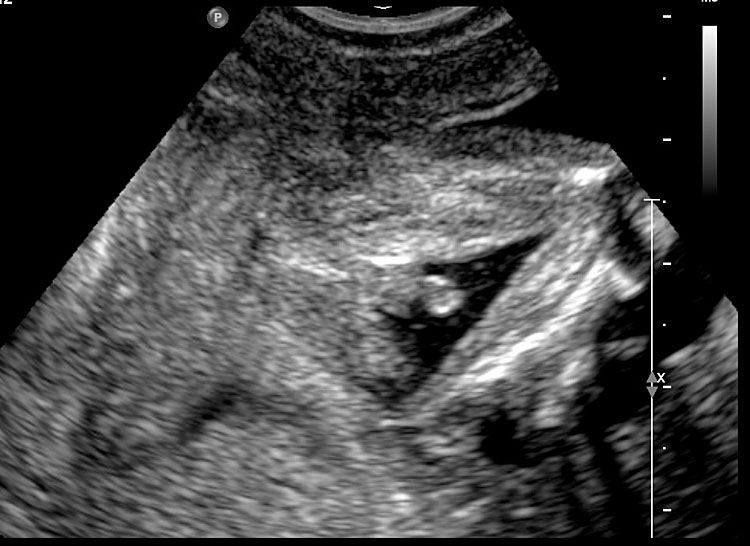
55
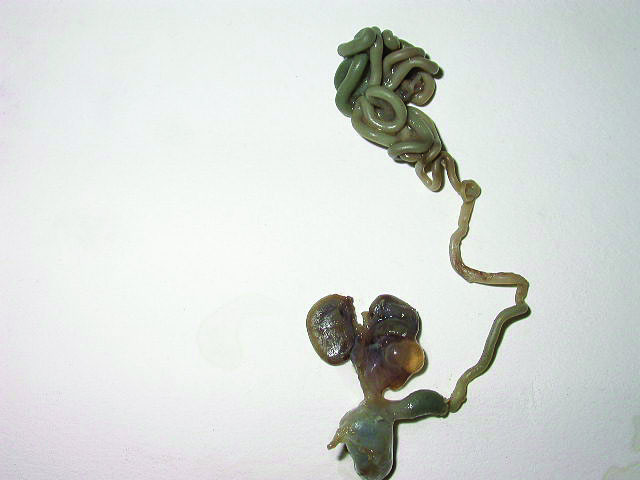
Fetal images of complete URSM sequence. (A) Coronal scan of fetal abdomen showing bilateral gross hydronephrosis (arrows) with a thick-walled conical cystic structure (arrow head) in pelvis with oligohydramnios. (B) Oblique scan of lower abdomen shows dilated distal colon (arrow) with echogenic calcified meconium. (C) Tangential scan of perineum showing nonvisualized anus. (D) Autopsy picture showing a common cloacal pouch to which the ureters and rectum are connected.
(A) |
(B) |
(C) |
(D) |
56
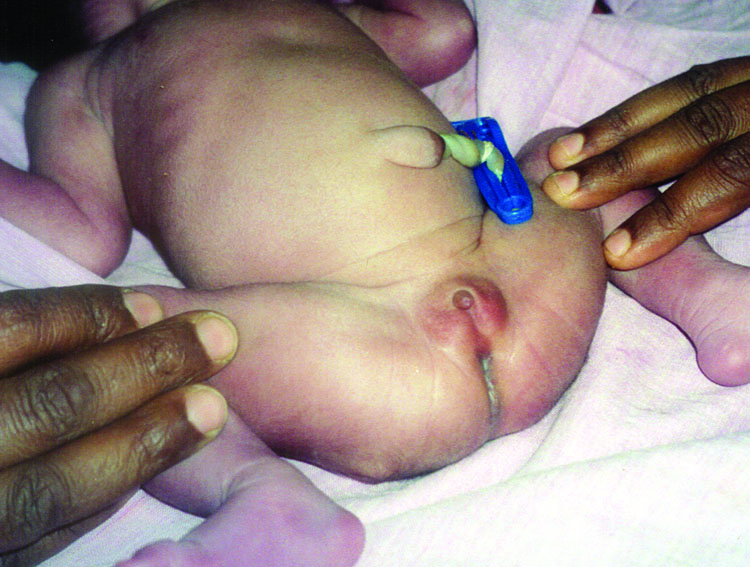
Partial URSM sequence. Coronal scans of fetal abdomen shows (A) gross hydronephrosis of right kidney and (B) agenesis of left kidney (arrow). (C) Scan of perineum shows nonvisualized anus. (D) Photograph of the newborn baby shows anal atresia.
(A) |
(B) |
(C) |
(D) |
57
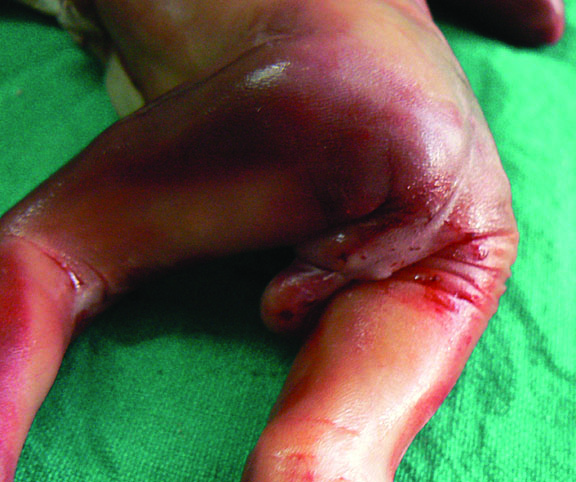
Partial URSM sequence. (A) Axial scan shows bilateral hydronephrosis. (B) Scan of pelvis shows urinary bladder (BL). (C) Scan of perineum shows an ambiguous genitalia and absent anus. (D) Photograph of the fetus confirms the findings.
- OEIS complex/cloacal anomaly (described in chapter on Ultrasound of the Fetal Abdominal Wall) (Figure 58).
- VACTERL association denotes the complex combination of anomalies of vertebrae, anus, cardiac, tracheoesophageal, renal and limb anomalies. Vertebral anomalies can vary from simple anomalies like hemivertebra (Figure 51), spinal dysraphism to gross kyphoscoliosis. Anorectal malformation can be low or high, cardiac anomalies can range from ventricular septal defect to complex anomalies. There can be esophageal atresia with or without tracheoesophageal fistula. Renal anomalies can be a spectrum from agenesis, to positional anomalies or complex obstructive lesion. Limb anomaly is usually a radial ray aplasia. When one component anomaly is seen careful search for other components must be performed. When a spinal anomaly or esophageal atresia is seen, looking for the fetal anus helps to categorize if it is isolated or part of VACTERL association.
- One of the features of anorectal malformation described earlier is dilated colon with echogenic meconium balls in it (Figures 54B and 59A). It is now known to be a nonspecific finding because of the large normal range of caliber of colon and echogenic meconium seen as a normal feature towards term. Therefore, seeing the fetal anus is crucial to diagnose if it is anorectal malformation or a normal finding (Figures 59 and 60).
- Bladder exstrophy is persistently non-visualized urinary bladder with normal amniotic fluid should raise the suspicion of bladder exstrophy. The redundant bladder mucosa is seen as a soft tissue mass at the lower abdominal wall inferior to cord insertion. It may be associated with anal atresia (Figure 61).
- Caudal dysplasia sequence (CDS) is characterized by a series of congenital abnormalities, including complete or partial agenesis of the sacrum and lumbar vertebrae associated with pelvic deformity. Femoral hypoplasia, clubbed feet and flexion contractures of the lower extremities are also commonly seen. Additionally, CDS is often associated with anomalies of the gastrointestinal and genitourinary tract and heart, as well as with neural tube defects. On prenatal sonography, the fetus with the severe form of caudal regression shows abrupt interruption of the spine at the dorsal or lumbar level and abnormal position of the lower limbs. The pelvic bone is small and fused with reduction in distance between femoral heads. The femur bones are fixed in a ‘V’ pattern, giving a typical ‘Buddha's poise’ (Figure 62). In the milder form of sacral agenesis there is complete or partial agenesis of sacrum (Figure 63). Both forms may be associated with anal atresia.
- Isolated anorectal malformation can be a low type of covered anus where the anal opening is not seen because of skin covering the anus. Here the anal sphincter complex is normally developed. Hence, the fetal anus will be seen on sonography which results in a false negative situation. In high anorectal malformation the rectum stops high above the perineum and so the normal anus is not seen.
(A) |
(B) |
(C) |
(D) |
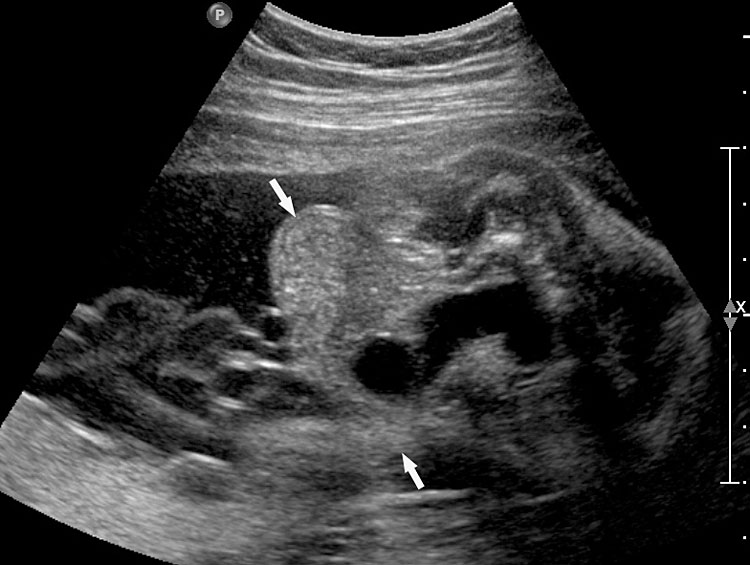
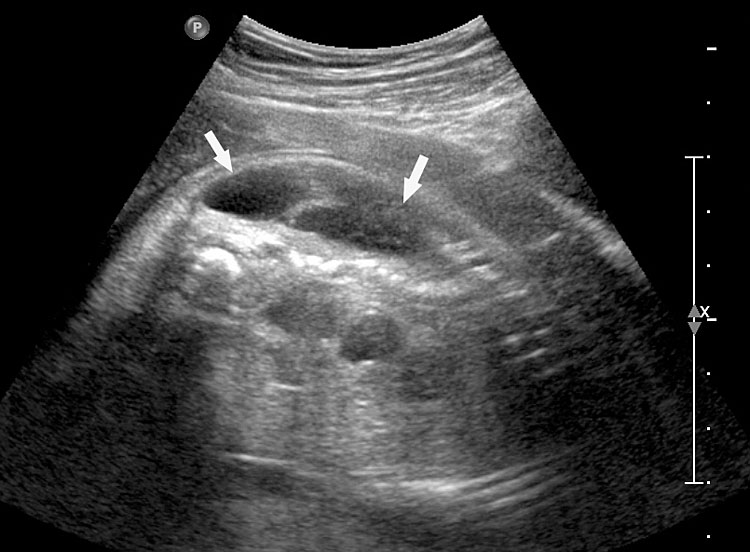
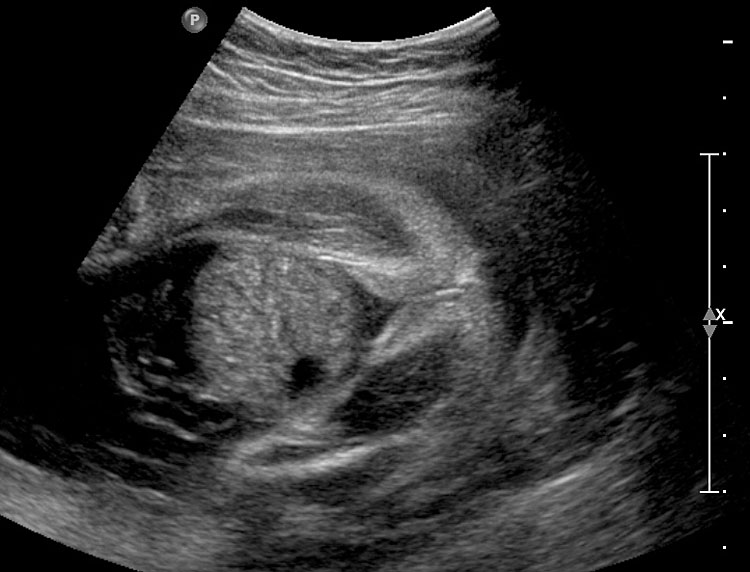
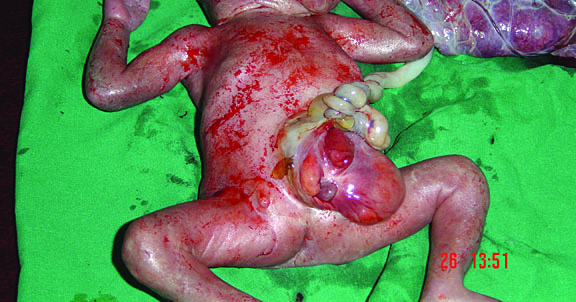
58
Fetus of OEIS complex. (A) Shows the omphalocele (arrows). (B) Shows the spinal anomaly with meningocele (arrows). (C) Scan of perineum shows absent anus. (D) Photo of fetus confirming the features.
(A) |
(B) |
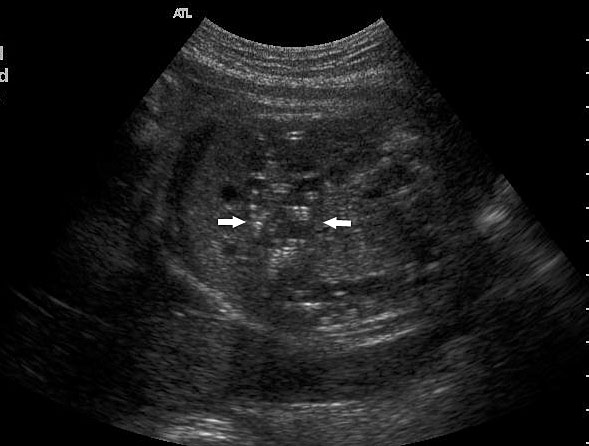
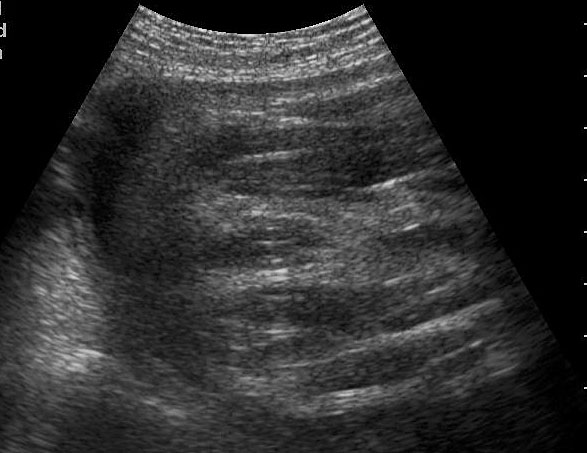
59
(A) Axial section showing the dilated colon with calcified meconium balls (arrows). (B) Scan of perineum showing absent anus indicating anal atresia.
(A) |
(B) |
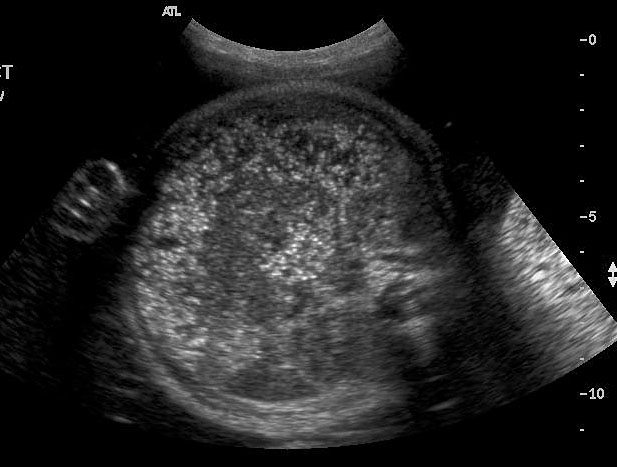
60
(A) Axial scan showing dilated colon with echogenic meconium balls. (B) Scan of perineum showing normal anus ruling out anal atresia.
(A) |
(B) |
(C) |
(D) |
(E) | |
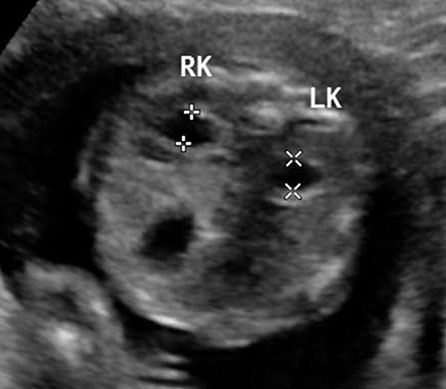
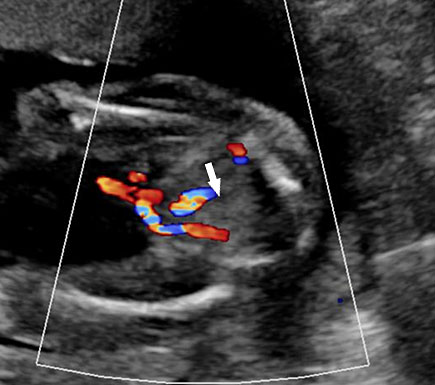
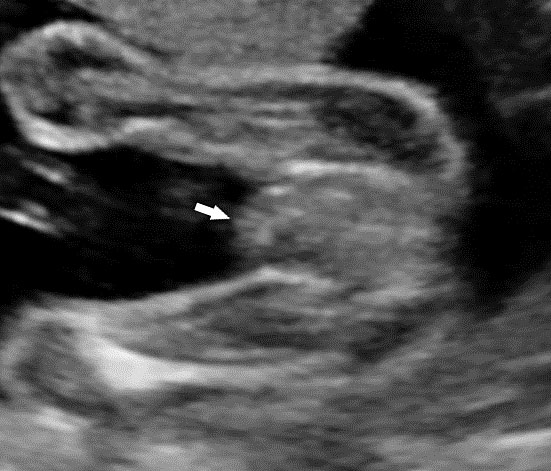
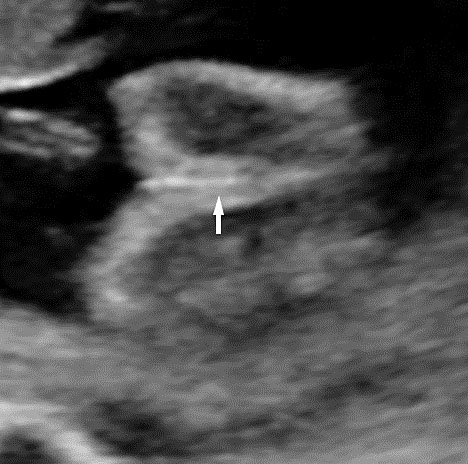
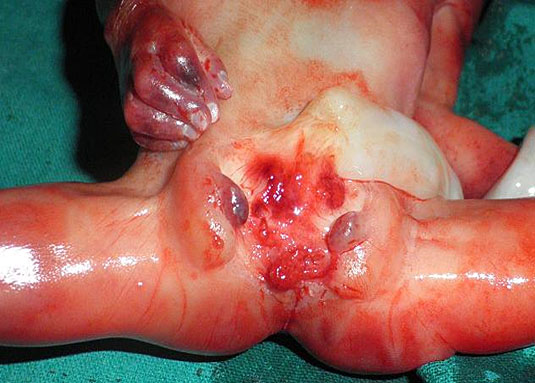
61
Bladder exstrophy. (A) Axial scan of fetal abdomen revealing bilateral mild hydronephrosis. (B) Color Doppler scan of pelvis showing non-visualized urinary bladder between the umbilical arteries (arrow). (C) Image shows the redundant bladder mucosa seen as a soft tissue mass (arrow) at the lower abdominal wall inferior to cord insertion. (D) Scan of perineum revealing non-visualized anus (arrow). (E) Photo of fetus confirming the features.
(A) |
(B) |
(C) |
(D) |
(E) | |
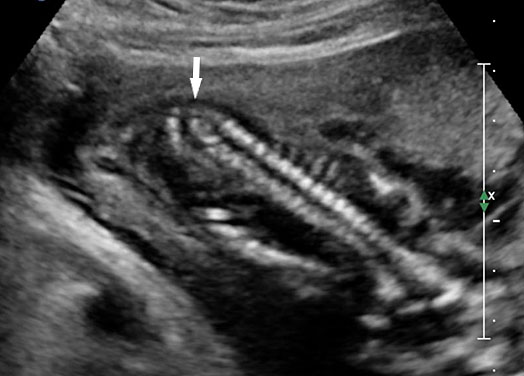
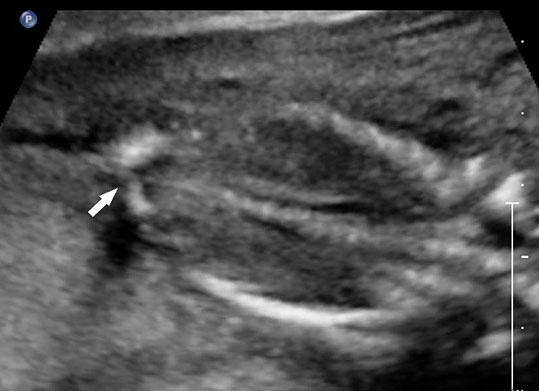
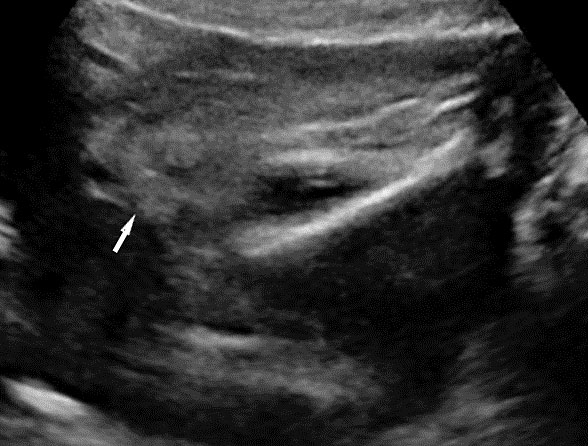
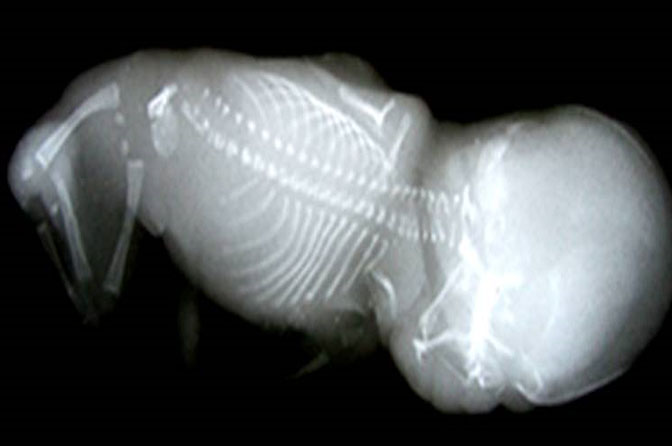
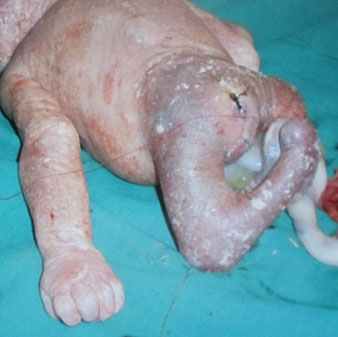
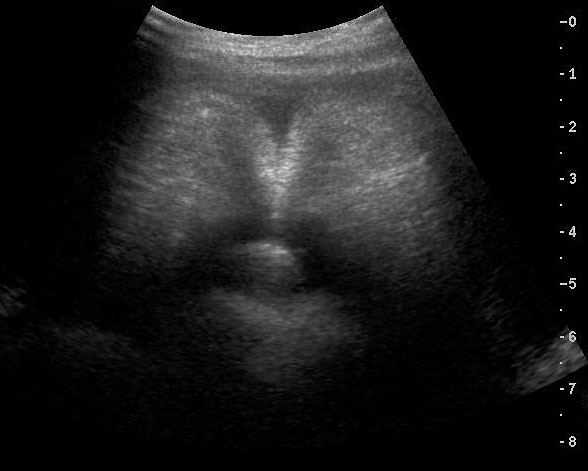
62
Caudal regression syndrome. (A) Longitudinal scan of fetal spine shows the abrupt termination (arrow) at the lumbar level. (B) Coronal scan demonstrating the small fused pelvic bone (arrow). (C) Scan of pelvis and perineum showing the non-visualized anus (arrow) and abnormal position of lower limbs. (D) Fetogram and (E) photo of fetus confirming the features.
(A) |
(B) |
(C) | |
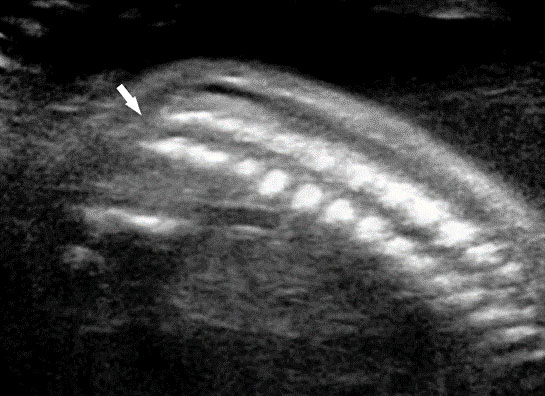
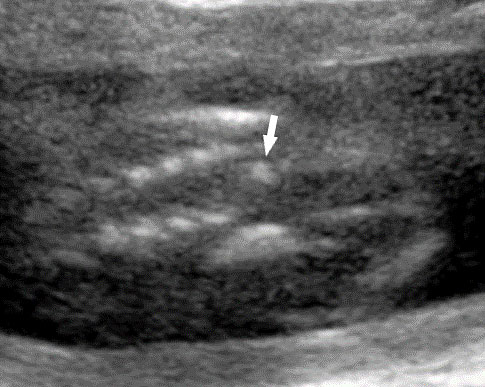
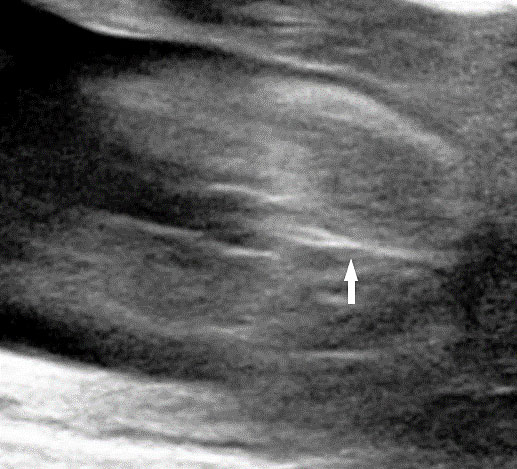
63
Sacral agenesis. (A) Longitudinal scan. (B) Coronal scan of fetal spine shows the non-visualized sacrum (arrow). (C) Scan of perineum revealing non-visualized anus.
PRACTICE RECOMMENDATIONS
- The screening of the fetal GIT can be achieved by the three axial sections obtained as part of second trimester anomaly scan (STAS) – namely upper and mid-abdomen, pelvis and a coronal scan of the abdomen.
- The high frequency linear transducer and EVS must be used to see better details of organs whenever possible.
- The degree of polyhydramnios in obstructive anomaly of the GIT decreases with the more distal the obstruction and increase with the gestation as swallowing and peristalsis become more frequent and vigorous.
- Direct visualization of esophagus is unnecessary.
- In esophageal atresia without TEF it is not necessary to look for pouch sign when the features are characteristic, namely, persistently nonvisualized stomach associated with polyhydramnios, which is not attributable to any other anomaly.
- Esophageal atresia with tracheoesophageal fistula can be diagnosed by looking for the pouch sign.
- Sonographic features of pyloric atresia are polyhydramnios, grossly dilated stomach with hyperperistalsis and dilated esophagus due to regurgitation caused by the distal obstruction.
- Sonographic sign of duodenal obstruction is “double bubble” sign with polyhydramnios.
- The characteristic feature of the gastrointestinal duplication cysts is the two layer sign or the bowel signature in the wall.
- There is a high risk of cystic fibrosis in fetuses presenting with the triad associating fetal hyperechogenic bowel, intestinal loop dilatation, and non-visualized gall bladder.
- The nonvisualization of anus is diagnostic of anorectal malformation.
- The nonvisualization of anus is seen in urorectal septal malformation (URSM) sequence and form part of complex associated anomalies like VACTERL association, caudal dysplasia, trisomy 18 and 21, OEIS complex or bladder exstrophy complex.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
REFERENCES
American Institute of Ultrasound in Medicine. AIUM practice guideline for the performance of obstetric ultrasound examinations. J Ultrasound Med 2010;29(1):157–66. doi: 10.7863/jum.2010.29.1.157. PMID: 20040792. | |
Salomon LJ, Alfirevic Z, Berghella V, et al. Practice guidelines for performance of the routine mid-trimester fetal ultrasound scan. Ultrasound Obstet Gynecol 2011;37(1):116–26. doi: 10.1002/uog.8831. PMID: 20842655. | |
Khurana A, Makhija B, Deka D, et al. Society of Fetal Medicine Practice Guidelines for the Second Trimester Anomalies Scan. Fetal Med 2014;1:11–15. https://doi.org/10.1007/s40556-014-0007-x. | |
Kassif E, Weissbach T, Kushnir A, et al. Esophageal atresia and tracheoesophageal fistula: prenatal sonographic manifestation from early to late pregnancy. Ultrasound Obstet Gynecol 2021;58(1):92–8. doi: 10.1002/uog.22050. PMID: 32304613. | |
Garabedian C, Sfeir R, Langlois C, et al. Does prenatal diagnosis modify neonatal treatment and early outcome of children with esophageal atresia? Am J Obstet Gynecol 2015;212(3):340.e1–7. doi: 10.1016/j.ajog.2014.09.030. Epub 2014 Sep 28. PMID: 25265404. | |
Vijayaraghavan SB. Antenatal diagnosis of esophageal atresia with tracheoesophageal fistula. J Ultrasound Med 1996;15(5):417–9. doi: 10.7863/jum.1996.15.5.417. PMID: 8731453. | |
Solt I, Rotmensch S, Bronshtein M. The esophageal 'pouch sign': a benign transient finding. PrenatDiagn 2010;30(9):845–8. doi: 10.1002/pd.2568. PMID: 20582925. | |
Choudhry M, Boyd PA, Chamberlain PF, et al. Prenatal diagnosis of tracheo-oesophageal fistula and oesophageal atresia. PrenatDiagn 2007;27(7):608–10. doi: 10.1002/pd.1745. PMID: 17457956. | |
Houben CH, Curry JI. Current status of prenatal diagnosis, operative management and outcome of esophageal atresia/tracheo-esophageal fistula. PrenatDiagn 2008;28(7):667–75. doi: 10.1002/pd.1938. PMID: 18302317. | |
Kalache KD, Chaoui R, Mau H, et al. The upper neck pouch sign: a prenatal sonographic marker for esophageal atresia. Ultrasound Obstet Gynecol 1998;11(2):138–40. doi: 10.1046/j.1469-0705.1998.11020138.x. PMID: 9549842. | |
Pardy C, D'Antonio F, Khalil A, et al. Prenatal detection of esophageal atresia: A systematic review and meta-analysis. ActaObstetGynecol Scand 2019;98(6):689–99. doi: 10.1111/aogs.13536. Epub 2019 Mar 6. PMID: 30659586. | |
Kansra M, Raman VS, Kishore K, et al. Congenital pyloric atresia – nine new cases: Single-center experience of the long-term follow-up and the lessons learnt over a decade. J Pediatr Surg 2018;53(11):2112–6. doi: 10.1016/j.jpedsurg.2018.04.015. Epub 2018 Apr 14. PMID: 29754879. | |
Lépinard C, Descamps P, Meneguzzi G, et al. Prenatal diagnosis of pyloric atresia-junctional epidermolysis bullosa syndrome in a fetus not known to be at risk. Prenat Diagn 2000;20(1):70–5. doi: 10.1002/(sici)1097-0223(200001)20:1<70::aid-pd747>3.0.co;2-e. PMID: 10701857. | |
Choudhry MS, Rahman N, Boyd P, et al. Duodenal atresia: associated anomalies, prenatal diagnosis and outcome. Pediatr Surg Int 2009;25(8):727–30. doi: 10.1007/s00383-009-2406-y. Epub 2009 Jun 24. PMID: 19551391. | |
Bishop JC, McCormick B, Johnson CT, et al. The Double Bubble Sign: Duodenal Atresia and Associated Genetic Etiologies. Fetal Diagn Ther 2020;47(2):98–103. doi: 10.1159/000500471. Epub 2019 Jun 5. PMID: 31167209; PMCID: PMC6893095. | |
Kimble RM, Harding JE, Kolbe A. Does gut atresia cause polyhydramnios? Pediatr Surg Int 1998;13(2–3):115–7. doi: 10.1007/s003830050262. PMID: 9563021. | |
Pierro A, Cozzi F, Colarossi G, et al. Does fetal gut obstruction cause hydramnios and growth retardation? J Pediatr Surg 1987;22(5):454–7. doi: 10.1016/s0022-3468(87)80269-5. PMID: 3295174. | |
Bittencourt DG, Barini R, Marba S, et al. Congenital duodenal obstruction: does prenatal diagnosis improve the outcome? Pediatr Surg Int 2004;20(8):582–5. doi: 10.1007/s00383-004-1235-2. Epub 2004 Aug 25. PMID: 15338169. | |
Saalabian K, Friedmacher F, Theilen TM, et al. Prenatal Detection of Congenital Duodenal Obstruction-Impact on Postnatal Care. Children (Basel) 2022;9(2):160. doi: 10.3390/children9020160. PMID: 35204881; PMCID: PMC8870145. | |
Virgone C, D'antonio F, Khalil A, et al. Accuracy of prenatal ultrasound in detecting jejunal and ileal atresia: systematic review and meta-analysis. Ultrasound Obstet Gynecol 2015;45(5):523–9. doi: 10.1002/uog.14651. PMID: 25157626. | |
Vijayaraghavan SB, Lalitha R, Jaleel Ahamed A. Antenatal diagnosis of Congenital Chloride diarrhea. Ind J Med Ultrasound 1998;4:17–8. | |
Dicke JM, Crane JP. Sonographically detected hyperechoic fetal bowel: significance and implications for pregnancy management. Obstet Gynecol 1992;80(5):778–82. PMID: 1407915. | |
Mekki C, Aissat A, Mirlesse V, et al. Prenatal Ultrasound Suspicion of Cystic Fibrosis in a Multiethnic Population: Is Extensive CFTR Genotyping Needed? Genes (Basel) 2021;12(5):670. doi: 10.3390/genes12050670. PMID: 33946859; PMCID: PMC8145404. | |
Kessels SJM, Carter D, Ellery B, et al. Prenatal genetic testing for cystic fibrosis: a systematic review of clinical effectiveness and an ethics review. Genet Med 2020;22(2):258–67. doi: 10.1038/s41436-019-0641-8. Epub 2019 Aug 30. PMID: 31467445. | |
de Becdelièvre A, Costa C, Jouannic JM, et al. Comprehensive description of CFTR genotypes and ultrasound patterns in 694 cases of fetal bowel anomalies: a revised strategy. Hum Genet 2011;129(4):387–96. doi: 10.1007/s00439-010-0933-1. Epub 2010 Dec 24. PMID: 21184098. | |
Vijayaraghavan SB, Prema AS, Suganyadevi P. Sonographic depiction of the fetal anus and its utility in the diagnosis of anorectal malformations. J Ultrasound Med 2011;30(1):37–45. doi: 10.7863/jum.2011.30.1.37. PMID: 21193703. | |
Escobar LF, Weaver DD, Bixler D, et al. Urorectal septum malformation sequence. Report of six cases and embryological analysis. Am J Dis Child 1987;141(9):1021–4. doi: 10.1001/archpedi.1987.04460090098038. PMID: 3618561. | |
Gupta A, Bischoff A, Peña A, et al. The great divide: septation and malformation of the cloaca, and its implications for surgeons. Pediatr Surg Int 2014;30(11):1089–95. doi: 10.1007/s00383-014-3593-8. Epub 2014 Sep 14. PMID: 25217828; PMCID: PMC4302733. |
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards programme CLICK HERE)