This chapter should be cited as follows:
Homfray T, Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.415603
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 4
Fetal development and maternal adaptation
Volume Editor: Professor Asma Khalil, The Royal College of Obstetricians and Gynaecologists, London, UK; Fetal Medicine Unit, Department of Obstetrics and Gynaecology, St George’s University Hospitals NHS Foundation Trust, London, UK

Chapter
Genetic Conditions: Chromosomal and Single Gene Disorders and Non-invasive Prenatal Testing
First published: February 2021
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

INTRODUCTION
Chromosome analysis can be undertaken by a number of different methods. The type of analysis undertaken is dependent on the indication for testing and the availability in different countries of different techniques. It continues to change, and the separation of cytogenetics and molecular genetics is no longer practical. The skills required to analyze the karyotype are being lost following the introduction of the more sensitive molecular techniques. There remains the need for the standard karyotype in certain circumstances.
Single gene disorders can increasingly be diagnosed during a pregnancy even in the absence of a family history, as sequencing of genes through next generation sequencing becomes quick enough to deliver results for management of a pregnancy.
Invasive prenatal diagnosis is much safer than previously but the preference by patients for non-Invasive testing is driving this technology forwards. This needs to be cautiously reviewed for the sensitivity and specificity of these tests.
CHROMOSOME ABNORMALITES
The karyotype (Figure 1)
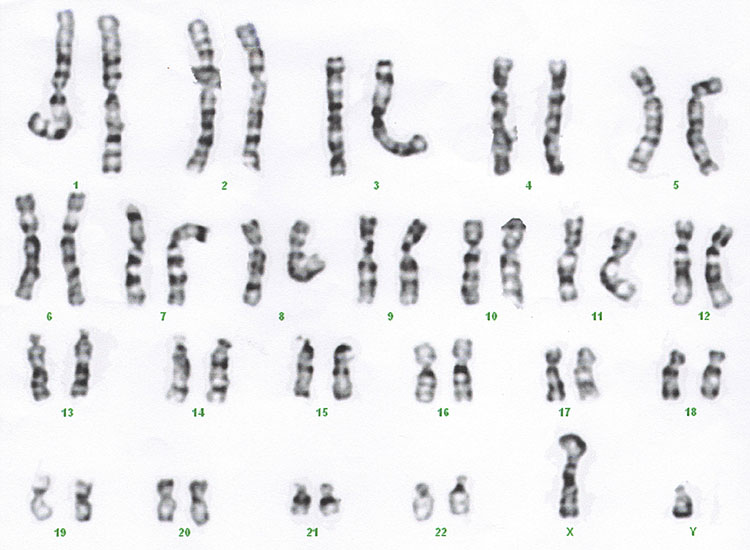
1
Standard karyotype.
Historically chromosomes were cultured and then the mitotic cells were arrested in metaphase where chromosomes are in their most condensed form and they can be examined down the microscope. The size of the abnormality that can be identified depends on the quality of the preparation and the part of the chromosome that is deleted or duplicated. Different tissues can be more difficult to get high quality preparations. The preparation from a chorionic villus sample is less good often than from an amniocentesis.
The result uses a nomenclature that describes the chromosome number, the arm of the chromosome where the abnormality is located if relevant and the band number that the abnormality is located.
Standard karyotype:
- Result available in 7–14 days, owing to the time required for the culture of cells.
- Can identify deletions of 5 Mb and duplications of 8 Mb, quality of preparation may vary.
QFPCR (quantitative fluorescence polymerase chain reaction) (Figure 2)
|
|

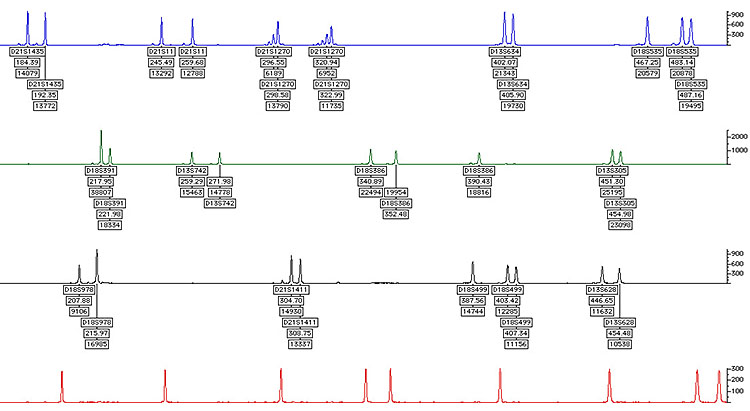
2
QFPCR.
Rapid analysis by DNA dosage of chromosome 13, 18 and 21 may look at X and Y, but may be more difficult to interpret, additional chromosomes occasionally may be added to this test, including 15, 16 and 22. This may only be added if the pregnancy has miscarried.
Array comparative genomic hybridization (aCGH) (Figure 3)
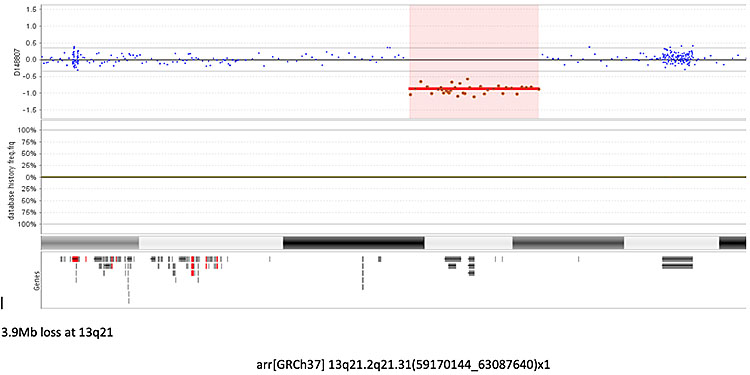
3
Array comparative genomic hybridization (aCGH) showing a microdeletion on chromosome 13, the red line represents the deleted probes in the fetal DNA, a green line above the blue line would represent a gain of probes or a duplication. Some areas of the genome are more gene rich than others and are covered by more probes. Each probe is represented by a single dot.
Array comparative genomic hybridization summary:
- DNA analysis.
- Dosage of DNA measured against a normal person.
- Identifies abnormalities down to about 100 Kb or less.
- Different arrays platforms will identify different size abnormalities.
- Nomenclature.
- Chromosome number followed by base pair abnormality from beginning of imbalance (base pair 1 is at the top of the P arm and highest number at bottom of Q arm) × 1 if deletion × 3 if duplication (except sex chromosome as males only have 1X).
- Only tells you about an imbalance.
- Will not indicate if the imbalance is attached to a different chromosome as would be the case with an unbalanced reciprocal chromosome translocation.
- Will not tell you if a trisomy is 20 to a Robertsonian Translocation only that there is an extra copy of the chromosome material.
- Will not identify Triploidy as all chromosome areas abnormal and therefore no obvious imbalance. (Triploidy would be identified on QFPCR.)
- Standard method of analysis in many units across the World and has replaced Karyotype analysis.
Fluorescent in situ hybridization (FISH) (Figure 4)
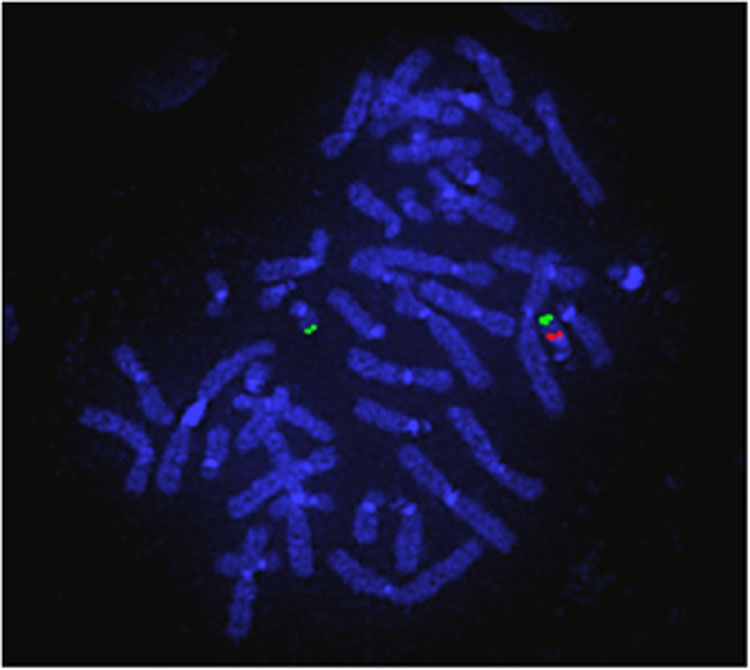
4
Fluorescent in situ hybridization (FISH). The 2 green probes represent the control probe to confirm the presence of both copies of the chromosome and the single red probe confirms that the other copy is missing. This is the 22q11.2 deletion confirming a diagnosis of DiGeorge syndrome.
A fluorescent probe is attached to a region of interest on a chromosome preparation. Normally it is used to answer a specific question such as Is there a deletion at Chromosome 22q11? Therefore this is not a screening test.
Types of chromosomal abnormalities
Trisomy – whole extra chromosome
Monosomy – missing whole extra chromosome
Deletion – missing part of a chromosome
Duplication – extra part of a chromosome
Inversion – part of chromosome inverted through 1800
Chromosome translocation – exchange of genetic material between two chromosomes when balanced all genetic material present and patient will be normal but at risk of having pregnancies with unbalanced karyotypes with chromosome deletions and duplications
Mosaicism – different cells with different chromosome number
Microdeletion/microduplication – not able to identify with standard karyotype
Common chromosomal abnormalities
TRISOMY
Down’s syndrome – trisomy 21
- Pregnancy:
- ↑ Nuchal translucency
- ↓ PAPPA (pregnancy associated plasma protein-A) +. ↓ ßhCG (human chorionic gonadotropin)
- Congenital heart disease atrioventricular septal defect (AVSD) most common
- Duodenal atresia (3rd trimester)
- Postnatal:
- Congenital heart disease, duodenal atresia
- Hypotonia, intellectual disability
- Characteristic facial features
- Wide sandal gap
- Other
Edward's syndrome – trisomy 18
- ↑ Nuchal translucency
- ↓ PAPPA + ↓ ßhCG
- Congenital heart disease
- Exomphalos
- Rocker bottom feet, clenched hands
- Lethal malformation syndrome, postnatally
Patau syndrome – trisomy 13
- Nuchal translucency
- ↓ PAPPA +. ↓ ßhCG
- Holoprosencephaly
- Anophthalmias
- Proboscis
- Cleft lip & palate
- Congenital heart disease
- Polydactyly
Other trisomies
Other abnormalities such as spina bifida may be present in trisomy 13 and trisomy 18.
No other trisomies can survive in non-mosaic form after birth, but may be identified at CVS and amniocentesis (see Mosaic abnormalities).
Certain other trisomies may be identified antenatally in their non-mosaic form, but either loose the extra copy of the trisomic chromosome or there will be an in utero or neonatal death. This is trisomic rescue.
- Trisomy 16 is the second commonest cause of miscarriage caused by a chromosome abnormality after monosomy X and may be present in 1.5% of conceptuses. Survival depends on loosing one of the chromosome 16s in the fetus. By trisomic rescue. Surviving fetuses have a high risk of severe in utero growth retardation.
- Other trisomies that may be identified antenatally include trisomy 8, 9 and 22.
Triploidy
69XXX or 69XXY
Whole extra set of chromosomes (69 total) (see Figure 8)
1% of all conceptions
10% of spontaneous abortions
- Paternal origin of extra chromosomes two-thirds (diandry)
- Result from dispermy
- Commonly result in spontaneous abortion
- Large placenta
- May develop partial mole
- Maternal origin of extra chromosomes one-third (digyny)
- Errors in meiosis II
- Early embryonic demise or late demise with small well formed fetus
- Small placenta
- Baby often small by 12 weeks and re-dated
- High risk of pre-eclampsia if pregnancy continues
MONOSOMY
Only surviving whole chromosome monsomy is 45X Turner’s syndrome.
Monosomy X
- NT >5 mm 99% spontaneous miscarriage in first or second trimester
- Many present with fetal hydrops and will not survive until 20 weeks
High proportion of surviving pregnancies mosaic with either 46XX or 46XY
Ultrasound features of Turner’s syndrome:
- High NT
- Coarctation of aorta
- Short long bones
- Rarely ambiguous Genitalia
Mosaic chromosome disorders
Chromosome mosaicism
The presence of more than one population of cells with different numbers of chromosomes.
Confined placental mosaicism
- May cause placental insufficiency and unexplained IUGR
- May lead to trisomic rescue if present at conception
- Trisomic rescue is where there is loss of one copy of a chromosome in an embryo that started with three copies of that chromosome, so that it now has only two copies
- May lead to imprinting disorders
- An imprinting disorder occurs when there is a parent of origin affect of a loss or gain of genetic material.
Trisomic rescue of chromosome 15 maternal disomy – Prader Willi, paternal disomy Angelman syndrome
- An imprinting disorder occurs when there is a parent of origin affect of a loss or gain of genetic material.
- Only certain chromosomes have been associated with imprinting disorders these are 6, 7, 11, 14, 15, 20, others may be found to be imprinted in the future
A baby may present with growth retardation in the second trimester of unknown cause. An amniocentesis may not reveal any chromosome abnormalities, but the fetus may have poor growth in view of placental chromosome abnormalities. Trisomy 16 is the most common trisomy associated with this.
True mosaicism
- Baby affected and has two different chromosome populations
- Variable affects depending on the number of cells with abnormal cell line
- Cannot predict affects on percentage of cells identified as variable between tissues
- Amniocytes more reliable to identify mosaicism than fetal blood
- Need to correlate result with ultrasound findings
- Well recognized chromosome mosaic syndrome
- Isochromosome 12p – Pallister Killian syndrome (see marker chromosome below)
- Mosaic trisomy 8
Other chromosome abnormalities
Deletions and duplications
These are termed microdeletions or microduplications if they cannot be identified on a standard karyotype and require other techniques for identification.
- Duplications are better tolerated by the fetus than a deletion.
- The effect of the duplication/deletion on the fetus will depend:
- On the size of the abnormality
- On the position of the abnormality with the number and function of the genes that are deleted or duplicated. If a deletion only deletes one gene but this is a critical gene, the baby can be much more seriously affected than a much larger deletion that does not carry any significant genes
- It is frequently important to look at the parental chromosomes to see if they carry the deletion/duplication as this might indicate if it is of significance to the fetal anomalies identified.
Many of these deletions/duplications are unique to that specific fetus, but even in those that are recurrent the features may be highly variable.
Common Microdeletion Syndromes and prenatal features (Table 1).
1
Recognized microdeletion syndromes (others recognized). All postnatally associated with other problems, all associated with variable degrees of intellectual disability.
Syndrome name | Deletion | Prenatal findings |
1p36del | CHD, SFD | |
Wolf Hiirschhorn 4p- | 4pter-variable | Cleft lip, SFD, microcephaly, CHD |
Cri du Chat 5p- | 5pter-variable | Small cerebellum SFD, CHD |
Williams | 7q11.23 | SFD, CHD supravalvar aortic stenosis |
8p23 del | CHD, diaphragmatic hernia | |
Kleefstra | 9q34.3 | Conotruncal heart defects |
Angelman/PraderWilli | 15q11.2 del | None |
Miller Dieker | 17p13.3 del | Lissencephaly, ventricular dilatation, s |
Koolan De Vries | 17q21.31 | SFD, Cleft lip & palate, CHD hydronephrosis |
Di George | 22q11.21 | CHD - Conontruncal defects |
18p deletion | 18pter-variable | SFD, talipes |
Cat eye | 22q11.1 dup | CHD - TAPVD, micrognathia, cleft palate |
Phelan McDermid | 22q13 del | Rarely macrocephaly |
CHD, congenital heart disease; SFD, small for dates; TAPVD, total anomalous pulmonary venous drainage.
Marker chromosome
A maker chromosome is an extra chromosome that is not a whole extra chromosome and can be a combination of parts of more than one chromosome.
- The origin of the marker chromosome can be identified by applying molecular probes.
- Many of these do not cause significant abnormalities, but others can lead to multiple abnormalities with a severe intellectual disability such as Isochromosome 12p leading to Pallister Killian Syndrome (Figure 5).
- Identifying the origin of the marker chromosome is essential to identify the severity of the problems for the baby.
- As marker chromosomes do not have a pair to undergo mitosis they are frequently lost during cell replication and, therefore, may be mosaic.
(a) |
(b) |


5
Chromosome mosaicism. Pallister Killian syndrome mosaic isochromosome 12p. (a) aCGH showing increased probes of 12p but not double the mount as the extra chromosome is not present in all cells. (b) Karyotype shows the extra two copies of 12p, again they will only be identified in some cells. A karyotype is required to clarify the abnormality.
Chromosome translocations
There are two types of chromosome translocations
- Robertsonian translocation (Figure 6)
- Balanced reciprocal translocation (Figure 7)
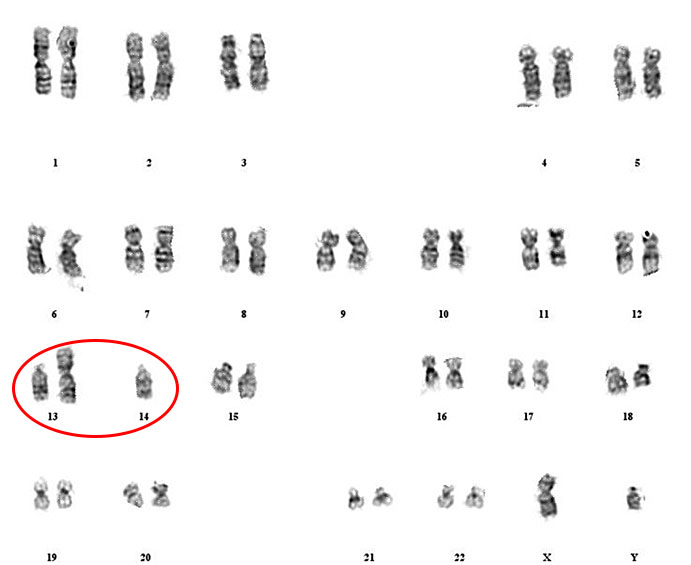
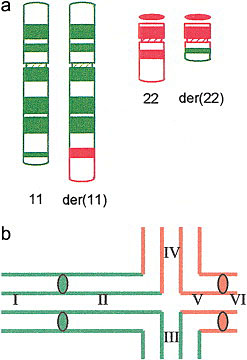
6
Robertsonian translocation. Balanced Robertsonian translocation resulting in a healthy person only having 45 chromosomes as one copy of chromosome 14 is attached to the top of another acrocentric chromosome in this case 13. The acrocentric chromosomes are more vulnerable to formation of these translocations and the short arms contain redundant copies of ribosomal DNA and therefore the loss of this does not affect the carrier.
(a) |
(b) |
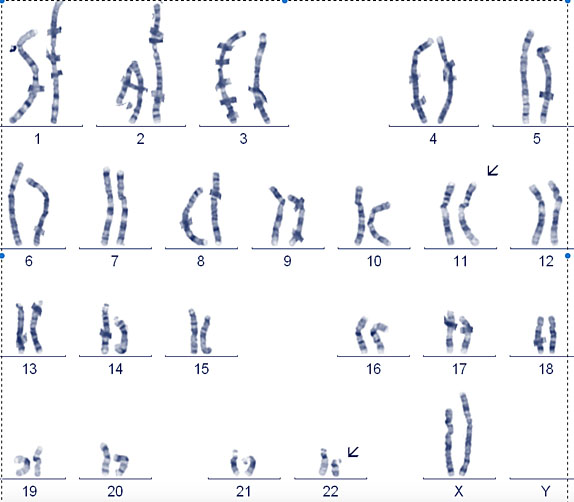
7
Balanced reciprocal translocation. 11 : 22 Balanced reciprocal translocation, this is the only recurring balanced reciprocal translocation, the others are almost always unique to a particular family. (a) Carrier is only at risk of problems if the translocation breakpoints are near or through important genes. This is a concern if it is a de novo event in a prenatal. (b) Shows an ideogram of the translocation and how the chromosomes align during meiosis.
The Robertsonian translocation:
- Involves those chromosomes known as the acrocentric chromosomes, 13, 14, 15, 21 & 22 which have very short (p) arms which are not essential as they contain multiple repetitive ribosomal RNA proteins which are redundant.
- The translocation results in the two long arms of the two acrocentric chromosomes joining together. Therefore, the carrier will only have a total of 45 chromosomes. Carriers of Robertsonian translocations have no effect from this.
- Carriers are at high risk of having a child with a major trisomy. 1 : 3000 people carrier a 13q14q Robertsonian translocation which can also cause infertility in some male carriers.
The balanced reciprocal translocation:
- Can involve any chromosomes and genetic material is exchanged between two unlike chromosomes.
- Only the 11 : 22 balanced reciprocal chromosome translocation seems to be a recurrent balanced reciprocal translocation with the others occurring as private translocations within a family.
- If the translocation is balanced, then the carrier of this should have no phenotypic affect from it.
- Occasionally a carrier of an apparently balanced translocation may have phenotypic affects as there is a microdeletion around the breakpoints of one of the chromosomes.
- A baby with a balanced translocation would be missed on an aCGH as this only shows imbalances of genetic material.
- Parental chromosomes are examined to see if a balanced translocation has arisen de novo. If de novo, aCGH would need to be performed to exclude a deletion/duplication.
- Balanced reciprocal translocations involving the X and Y chromosomes should be referred for an expert opinion as they may have different effects on the developing fetus to those involving the other chromosomes in view of X inactivation.
- The risk of a baby receiving an unbalanced karyotype from a parental balanced reciprocal translocation depends on the size of the translocated segments. Larger translocations are more likely to result in larger imbalances and, therefore, miscarriages are commoner than in the smaller translocations where the baby may be viable.
Single gene disorders
Inheritance patterns (Figure 8)
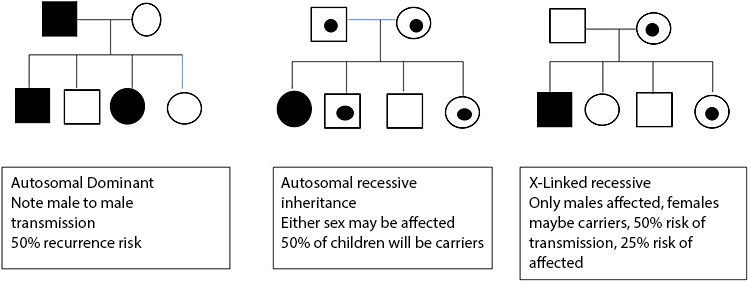
8
Inheritance patterns.
A disorder that occurs secondary to a mistake within a single gene is a single gene disorder.
- This can be inherited directly from an affected parent – autosomal dominant inheritance
- Huntington’s chorea, neurofibromatosis type 1
- Inherited from both unaffected parents – autosomal recessive inheritance
- Cystic fibrosis, sickle cell disease
- An unaffected mother may have an affected boy due to a mutation on the X chromosome – X linked recessive Inheritance
- Duchenne muscular dystrophy, Hunter’s syndrome
- A mutation can occur as a new event in a baby – de novo dominant inheritance
- Cornelia De Lange syndrome, Rubenstein Tabyii syndrome
- A mutation on the X chromosome occurring in a girl that does not cause a known disease in males normally due to early lethality, many are de novo
- Rett syndrome, incontinentia pigmenti
- Genes that are imprinted only cause a disease if they are inherited from one parent as expression of some genes is dependent on the parent of origin
- Angelman syndrome UEB3A mutation – maternal inheritance
- Beckwith Weidemann Syndrome, CDKNC1 – maternal inheritance
The effect of a mutation within a single gene will depend on many factors. As it is possible to undertake much more extensive testing now in the absence of a family history and a known mutation identified in a family member interpretation of genetic results needs to be undertaken by an expert that understands the complexity of the analysis.
GENETIC TESTING IN PREGNANCY
A genetic diagnosis may be suspected by the ultrasound findings but only rarely are these so specific that confirmation is not required by a separate test.
- In couples who have had a previous affected baby with typical ultrasound features, the diagnosis can be confirmed by ultrasound and decision about the pregnancy management does not need to wait until other tests are available.
- Many abnormalities are so severe that the prognosis of the pregnancy will not be altered by the underlying cause of the abnormalities and once again it is not necessary to await confirmation of the cause of the abnormalities. The etiology of the abnormalities is important for management of future at risk pregnancies.
Genetic testing may be undertaken after identification of a familial mutation, it is essential that the report of the familial mutation is available prior to undertaking testing.
An increasing number of couples are having pre-pregnancy screening for genetic disease in the absence of a family history of the disease. This normally involves testing a panel of genes for autosomal recessive and x-linked recessive diseases. Confirmation of these mutations in an accredited laboratory is required for prenatal diagnosis.
Non-invasive prenatal genetic testing (NIPT) (Figure 9)
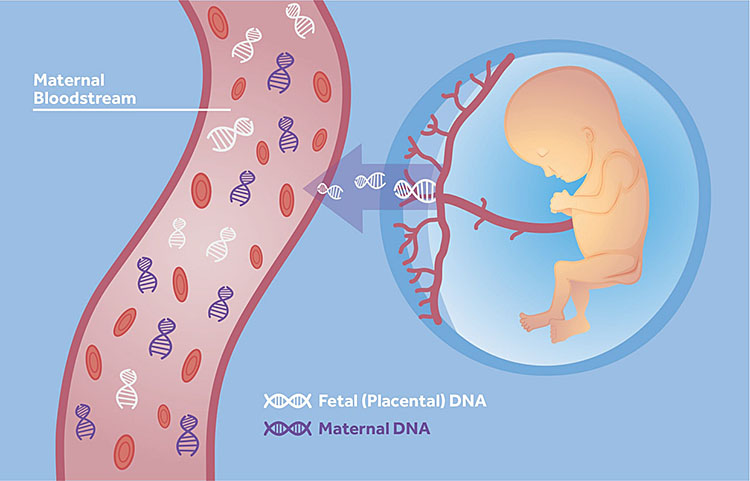
9
Placental DNA for non-invasive prenatal genetic testing.
Testing for fetal genetic abnormalities can now be undertaken from a maternal blood sample from 10 weeks' gestation.
Placental DNA is shed in the maternal circulation as free DNA. This DNA is not in the cells but is free in the circulation and is the result of DNA produced as a result of apoptosis of placental cells. The percentage of placental DNA in the maternal circulation increases with gestation.
Indications for NIPT:
- Screening for the major trisomies
- Screening for other microdeletion syndromes
- Screening for Rhesus disease in Rhesus negative mothers
- Identifying the sex of the fetus
- Testing for known mutations for specific ultrasound abnormalities, e.g. FGRFR3 in achondroplasia
- Diagnosis of known genetic mutations within a family as an alternative to invasive prenatal diagnosis.
Screening for the major trisomies
- NIPT for the major trisomies is a well established test which is highly accurate for trisomy 21 (detection rate (DR) >99%), trisomy 18 (DR 98%) and trisomy 13 (DR 99%) with a combined false positive rate (FPR) of 0.13%. Fetal sexing as part of the aneuploidy screening is performed but with a lower accuracy than for the major trisomies, different tests perform with different accuracies depending on the method of analysis. Sex chromosome abnormalities are not reliably detected by these tests.
- All abnormal results should be confirmed by an invasive test, as mosaicism will be missed by these tests.
- If there is an ultrasound abnormality including a high nuchal translucency >3 mm, then a CVS could be undertaken to confirm the abnormality. Confirmation by QFPCR is enough to proceed with termination of pregnancy if that is the desired option of the patient.
- If there are no ultrasound abnormalities, then an amniocentesis should be performed as this avoids the possibility of identifying confined placental mosaicism.
Screening for other microdeletions syndromes
- None of these tests have as yet been proven to have sufficient accuracy to recommend their use. This may change with time.
Screening for Rhesus disease in Rhesus negative mothers
- All Rhesus negative mothers are given anti D during the pregnancy to prevent Rhesus disease of the baby. This is obviously unnecessary if the baby is also Rhesus negative therefore genotyping the baby will prevent the use of anti D which is a blood product unnecessarily.
Identifying the sex of the fetus
- Babies at risk of X-linked disorders will only be at risk if they are male in X-linked recessive diseases and female in X-linked dominant disease. Invasive testing will only then be required for the sex at risk baby, hence undertaking this test prior to a CVS.
Testing for known mutations with ultrasound abnormalities
- Confirmation of a few diseases can be reliably made by NIPT as there are only a very few mutations that cause these abnormalities. This test is then reliable and may help pregnancy management. At present his includes FGFR3 for thanatophoric and achondroplasia and FGFR2 for Apert syndrome.
Diagnosis of known genetic mutations within a family as an alternative to invasive prenatal diagnosis (NIPD)
- A number of known single gene disorders such as cystic fibrosis and spinal muscular atrophy can be identified through NIPD. These tests need to be developed for each couple undergoing testing. It is possible to develop these tests for all single gene disorders except for those where the mother is a mosaic carrier for the disease in question. Identification of maternal mutations is more difficult in view of the presence of the mutation within the maternal circulation but tests using dosage of the mutation continue to be developed.
- The accuracy of NIPD will be less than for standard prenatal invasive diagnosis and as the miscarriage risk for these tests is so low in expert hands it is debatable whether this test is a better option than standard testing. As many are performed to decide if a pregnancy will continue this is a very important consideration.
Invasive prenatal testing
- Chorionic villus testing (CVS)
- Amniocentesis
- Fetal blood sampling (FBS)
- Other samples may be used such as fluid from drainage of fetal ascites or pleural effusion.
DNA can be extracted from all samples and the karyotype can be examined from all samples. It is important to remember the following when considering which sample to collect.
- The most DNA is available from the chorionic villi.
- The karyotype preparation is often better and smaller abnormalities can be identified from amniocytes. As most abnormalities are now identified on aCGH, this is now less important.
- Confined placental mosaicism (CPM) may be present and, therefore, an abnormality present in the chorionic villi may not be present in the fetus. This relates to chromosome abnormalities and normally mosaicism can be identified on the results which would indicate a second invasive test needs to be undertaken.
- Exome sequencing is a test that can look at a large number of genes at one time, this requires considerable amount of DNA and therefore a CVS is preferable but there are a few conditions where the fetus but not the placenta may be mosaic.
- More DNA can be extracted following amniocyte culture but this will delay the result.
PRACTICE RECOMMENDATIONS
- Diagnosis of a genetic disease during pregnancy can be confirmed following invasive and non-invasive prenatal testing, but invasive testing is required for confirmation of the diagnosis.
- Analysis of genetic disorders by amniocentesis in the absence of a known genetic disorder is often better than by chorionic villus sampling, but this has the disadvantage of only being possible from 16 weeks' gestation and the amount od DNA extracted may be small which may delay a definitive diagnosis and, therefore, it is necessary to decide which test should be undertaken to limit the delay in getting a result.
- If prenatal diagnosis is undertaken for a known genetic disorder, the results from the familial testing must be available to the laboratory for accurate prenatal diagnosis to be undertaken.
- Interpretation of results can only be made with full clinical information of the ultrasound abnormalities and therefore these must be provided to the laboratory.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
FURTHER RESOURCES
- Mckinlay Gardner RJ, Amor DJ. Gardner and Sutherland’s Chromosome Abnormalities and Genetic Counseling, 5th edn. Oxford Monographs on Medical Genetics, 2018.
- Firth HV, Hurst JA. Clinical Genetics & Genomics (Oxford Desk Reference), 2nd edn. Oxford University Press, 2017.
- UNIQUE Understanding Rare Chromosome and Gene Disorders: www.rarechromo.org.
- Online Inheritance in Man: www.omim.org.
- Phenotype browser: decipher.sanger.ac.uk.
- Gene Reviews: www.ncbi.nhm.nih.gov.
- Samura O. Update on noninvasive prenatal testing: A review based on current worldwide research. J Obstet Gynaecol Res 2020;8:11246–56.
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards programme CLICK HERE)