Cytogenetics
Authors
INTRODUCTION
Cytogenetics is the branch of genetics that studies the structure and behavior of chromosomes and their relation to human disease and disease processes. During the past three decades, the importance of clinical cytogenetics to the practice of obstetrics and gynecology has dramatically increased because clinical cytogenetics has a direct effect on the diagnosis, management, and prevention of many disorders that are caused by chromosome aberrations. For many chromosome disorders, physicians face medicolegal responsibilities in the areas of counseling, screening, and diagnosis, and obstetricians and gynecologists therefore must have knowledge about the human chromosome constitution and be able to apply basic principles of chromosome behavior to clinical practice. This chapter reviews important concepts and developments in cytogenetics and highlights their applications in the practice of obstetrics and gynecology.
SOURCES OF SPECIMENS FOR CHROMOSOME ANALYSIS
Chromosomal analysis can be performed on cells derived from many sources, including peripheral blood lymphocytes, fetal blood, oocytes and spermatozoa, blastomeres of embryos, chorionic villi, amniotic fluid cells, skin, bone marrow, fetal tissues (e.g. lung, liver), solid tumors, and ascites.
The first step in a chromosome analysis requires appropriate collection of the specimen. For example, peripheral blood and fetal blood samples must be placed in tubes containing sodium heparin to prevent clotting, whereas fetal tissues should be placed in sterile, balanced salt solutions to prevent dehydration. These suggestions may appear obvious and mundane, but a critically important chromosome analysis often cannot be performed because the specimen is contaminated, placed in a fixative (e.g. formalin), or allowed to dehydrate.
Specimen preparation varies with the type of tissue undergoing chromosome analysis (Table 1). In most instances, the tissues are cultured in a synthetic media supplemented with fetal calf serum, antibiotics, and glutamine to increase the number of cells in mitosis available for cytogenetic analysis. The time required to complete the chromosomal analysis also varies with the type of tissue (Table 1). In the case of prenatal diagnosis, the presence of actively dividing cells of the cytotrophoblast layer of the chorionic villi permits direct and immediate chromosome analysis, whereas the mesenchyme core of the chorion and amniotic fluid cells usually requires 6 to 14 days of tissue culture before harvesting for chromosome analysis. To evaluate infertility and repeated pregnancy loss, as well as newborns with birth defects, peripheral blood cells are commonly used in chromosome analysis because culturing of lymphocytes requires only 48 to 72 hours. For the diagnosis of hematologic disorders, particularly different types of leukemia, bone marrow cells rather than peripheral blood lymphocytes must be studied by direct preparation and after short-term tissue culture (24 to 48 hours). For skin, fetal and gonadal tissues, fibroblast cultures are established routinely. They usually require a minimum of 8 to 15 days to obtain a sufficient pool of dividing cells to permit cytogenetic analysis.
TABLE 1. Source of Tissue, Cell Type, and Applications for Cytogenetic Analysis
Source of Tissue | Cell Type | Applications |
Peripheral blood | T lymphocytes | Routine analysis |
Chorionic villi | Trophoblast | First-trimester prenatal diagnosis |
Amniotic fluid | Amniocytes | Second-trimester prenatal diagnosis |
Percutaneous umbilical blood | T lymphocytes | Second-trimester fetal diagnosis |
Fetal tissues | Fibroblast | Confirmation of diagnosis |
Bone marrow | White blood cells | Leukemia |
Fetal ovary | Oocytes | Meiosis |
Spermatozoa | Spermatozoa | Meiosis |
Solid tumors and ascites | Tissue Specific | Diagnosis, treatment, and prognosis |
Oocyte | Polar body | Preimplantation genetic diagnosis |
Embryo | Blastomere | Preimplantation genetic diagnosis |
Cells in tissue culture are prepared for chromosome analysis by the sequential addition of Colcemid®, which prevents spindle fiber formation, thereby arresting cells in the metaphase stage of mitosis; hypotonic solution (water or 0.7 M potassium chloride), which causes water to enter the cell, swelling the nuclei and separating chromosomes from one another so that they can be studied effectively after staining; fixative solution, usually a combination of methanol and acetic acid, which preserves the chromosomes permanently; and staining solutions that produce various banding patterns along the chromosome arms that enable each chromosome to be unequivocally identified. The most common staining treatments involve exposing the cells to an enzyme (trypsin or pancreatin), followed by Giemsa stain to produce G bands. Cells cultured in suspension (e.g. lymphocytes, ascites, bone marrow cells) can be processed by addition of Colcemid® and hypotonic solution to the culture media, followed by fixative, and, before staining, transfer from solution to a microscope slide. For tissue culture, cells can be grown attached to the surface of coverslips or directly on the microscope slide and processed completely in situ. The in situ method provides a relatively accurate assessment of how many independent cells from the original source have been analyzed, assuming that each colony was derived from one cell or, at most, a few cells.
THE HUMAN CHROMOSOME COMPLEMENT
One complete set of human chromosomes consists of 23 different kinds of chromosomes. A complete set is referred to as the haploid number (N); for humans, N = 23. Mature gametes, oocytes, and spermatozoa normally have haploid sets of chromosomes. A normal human somatic cell, regardless of origin, contains two complete sets of chromosomes, one of maternal origin and the other of paternal origin. There are 46 chromosomes in the nuclei of all human somatic cells; this number is known as the diploid number (2N); for humans, 2N = 46. Exceptions exist; mature red blood cells have no nuclei, whereas liver cells, chorionic villi on occasion, and some amniotic fluid cells may have four sets of chromosomes (i.e. 4N = 92).
Chromosomes are classified as autosomes or as sex chromosomes. There are 22 autosomes in the human chromosome complement. Because a somatic cell contains two members of each kind of chromosome, there are 22 homologous (morphologically similar) pairs in human cells. Cells with two X chromosomes are classified as female, and cells with one X and one Y chromosome are classified as male. These two chromosomes are the sex chromosomes. During gametogenesis, the paired condition becomes unpaired, so each parent usually contributes one member of each of the 23 pairs of chromosomes to a gamete. A set consisting of 23 different chromosomes is transmitted to the next generation by oocytes and spermatozoa. The only difference is that spermatozoa contain an X chromosome or a Y chromosome.
Chromosome morphology before 1970 was limited to the relative length and the position of the centromere. The centromere, or primary constriction, is the site of attachment of the spindle fibers essential for the orderly segregation of chromatids (duplicates of each chromosome) into daughter cells during mitosis and meiosis. At the metaphase of mitosis, chromosomes are already duplicated and the duplicates are held together by the centromere. The chromosome number of a cell is actually a count of the number of centromeres. When the centromere divides, each chromatid with its own centromere becomes a chromosome. The location of the centromere divides each chromosome into a short arm, designated p for petite, and a long arm, designated q. Chromosomes are metacentric if the centromere is located in the middle of the chromosome (i.e. the arms are of approximately equal length), submetacentric when the centromere is located away from the center, and acrocentric when the centromere lies close to one end of a chromosome. The ends of the short arms of acrocentric chromosomes often exhibit satellites, which are portions of the ends of chromosomes that seemingly are separated from the main body and apparently represent an altered coiling pattern of the chromatin. These satellite regions are associated with nucleolar formation. An acentric chromosome is one without a centromere and, lacking a site for spindle fiber attachment, is not passed to the daughter cells of the next cell generation. A dicentric chromosome has two centromeres that can produce a cycle of chromosome breakage if the two centromeres simultaneously move toward different daughter cells during cell division.
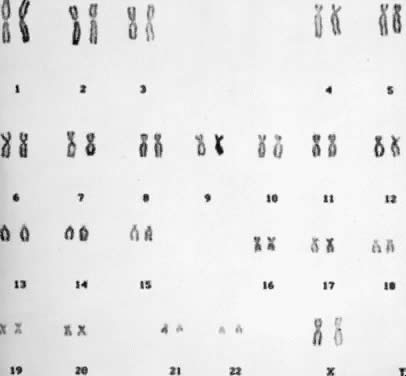
Staining procedures before 1970 produced chromosomes with limited morphologic variation. Consequently, individual chromosomes could not always be paired, nor could certain pairs be readily distinguished from other pairs. The Denver system3 of classification, which was named for the city where cytogeneticists met in 1961, grouped chromosomes into seven classes, as follows: chromosomes 1, 2, and 3 were group A; chromosomes 4 and 5 were group B; chromosomes 6 through 12 and the X chromosome were group C; chromosomes 13, 14, and 15 were group D; chromosomes 16, 17, and 18 were group E; chromosomes 19 and 20 were group F; and chromosomes 21 and 22 and the Y chromosome were group G. This system recognized that the staining techniques available at the time produced a homogeneous appearance along the entire length of the chromosome (Fig. 1). For example, the X chromosome was one of eight pairs of chromosomes in group C that could not be distinguished from each other.
|
In 1970, Caspersson and colleagues4 described a fluorescent staining technique that produced patterns of horizontal bands (Q bands, named after the compound quinacrine) that distinguished each of the 22 pairs of autosome chromosomes and the X and Y chromosomes. This development was a major advancement in the field of cytogenetics. Previously established chromosome syndromes were confirmed. More refined analysis of chromosome rearrangements was possible because the resolving power for detection of chromosome aberrations was greatly enhanced. New chromosome syndromes were identified. Cancer cytogenetics progressed rapidly, leading to the identification of chromosome aberrations associated with specific hemopoietic malignancies. Assigning of genes to specific chromosome sites (gene mapping) became possible.
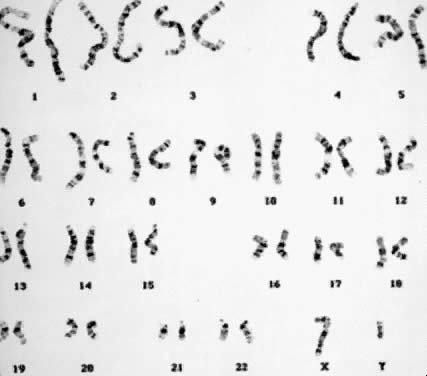
Several other banding techniques were developed shortly afterward that did not require the use of a fluorescent microscope.5,6 These included trypsin digest of chromosomes followed by Giemsa staining (G banding), currently the most widely used of banding techniques (Fig. 2);7 and, R banding,8 in which exposure to salts solutions at high temperature produces a banding pattern that is opposite that of Q banding and G banding. For example, where Q banding shows bright fluorescence and G banding dark bands, R bands are light, and where Q banding shows dull fluorescence and G banding light bands, R bands are dark. Additional techniques have also been developed with special qualities to identify unique-appearing chromosomes. For example, C banding9 delineates each chromosome in regard to the location and number of centromeric regions. Silver staining,10 together with Q-band profiles, identifies the parental origin and the stage of meiotic nondisjunction in human trisomies of acrocentric chromosomes.
|

Colcemid® inhibits spindle fiber formation, preventing chromosome movement to the poles of the cell. Chromosomes that are exposed to Colcemid® for more than 1 to 2 hours also show increased spiralization (compaction) of their arms. With standardized harvesting procedures, including a 1- to 2-hour exposure to Colcemid®, chromosomes usually show 300 to 400 bands in the total chromosome complement. By shortening the time of Colcemid® exposure (e.g. to 15 minutes) and by analyzing stages earlier than metaphase (the stage of maximum chromosome contraction), the technique of high-resolution banding made possible the identification of a much larger number of bands, up to 1200 bands in the total complement (Fig. 3).11,12 High-resolution banding is particularly important in detecting extremely small deletions, such as a deletion in the proximal portion of the long arm of chromosome 15 associated with Prader-Willi syndrome,13 the deletion in chromosome 11 associated with Wilms-aniridia syndrome,14 the deletion of a single chromosome band in the long arm of chromosome 13 associated with retinoblastoma,15 and a deletion in the X chromosome associated with four different genetic syndromes and that contributed to the precise gene mapping and cloning of the gene for Duchenne muscular dystrophy and the eventual detection of the protein, dystrophin, which is lacking in affected males.16
|
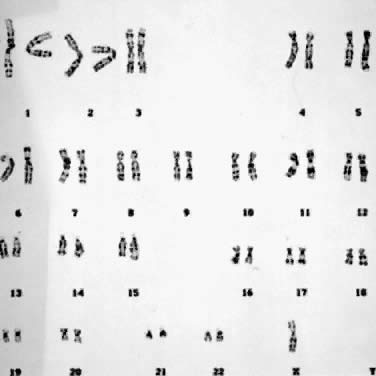
With the rapid expansion of the application of banding techniques, there was a need to refine the classification and identification of individual chromosomes. This need was addressed by the Paris nomenclature.5 Numeric designations were assigned to each chromosome, and more importantly, regions and subregions were given numbers. This provided one numeric language system by which cytogeneticists could communicate with each other. For example, the designation, 46,XX,del(5)(p21), indicates a female cell with a deletion of band 21 in the short arm of chromosome 5 (Fig. 4); the designation 47,XY,+ 21 indicates a male cell with 47 chromosomes because of the presence of an extra number 21 chromosome (i.e., trisomy 21) (Fig. 5); and, 45,X/46,XY indicates a mosaic with two cell lines, one with a single X and the other with an X chromosome and a Y chromosome.
|
|
CYTOGENETIC ANALYSIS
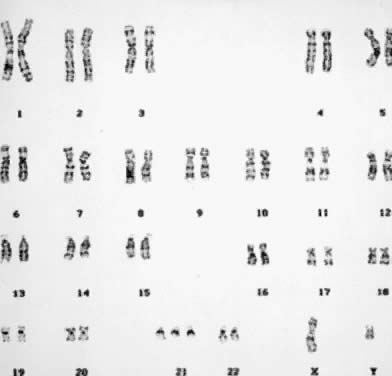
A routine cytogenetic analysis involves evaluating 15 to 20 cells to determine their modal chromosome number and assessing the structural integrity of each chromosome in the complement. The modal chromosome number can be determined by counting chromosomes through the microscope. Identification of an extra or missing chromosome is possible by microscopic examination, and the finding is confirmed through the preparation of a set of karyotypes. A karyotype is a photographic or computer-generated representation of the chromosomes in a cell arranged according to size, centromeric position, and banding pattern. Assessment of the structural integrity of each chromosome in the complement can also be made through the microscope, and gross structural chromosome aberrations can be detected as well. However, a set of two or more karyotypes always must be prepared. Cells with well-prepared chromosomes that exhibit a total of 350 to 400 bands in the complement are considered an essential part of any routine cytogenetic analysis (Fig. 2). A standard chromosome analysis, therefore, requires that the final interpretation be based on a chromosome count of at least 15 to 20 cells, the preparation and careful evaluation of two to three karyotypes consisting of well-banded chromosomes, and the exclusion of any structural chromosome aberrations. Subtle chromosome aberrations, such as the gain or loss of a single band, usually are detected as the result of a detailed analysis of the banding pattern of chromosomes magnified thousands of times by means of photographic or computer processing. In this way, homologous chromosomes can be examined meticulously and compared for small but clinically significant structural differences. The number of cells analyzed varies according to the reason for referral, the initial cytogenetic findings, the protocol of each laboratory, and the quality of the chromosome morphology.
If an abnormal chromosomal complement is present in all of the cells of a specimen, analysis of a small number of cells10,11,12 may be sufficient for diagnostic purposes. The presence of a single cell with a chromosome aberration usually is attributed to an error in cell division during tissue culture or a technical artifact arising in the course of harvesting. In most cases, the parents are informed that such a finding is not associated with anomalous fetal development. Nevertheless, the presence of an abnormal cell may necessitate analyzing 20 to 50 or more cells to distinguish between true mosaicism, which could be responsible for fetal maldevelopment, and pseudomosaicism (false mosaicism) caused by technical artifact or an in vitro error.
True chromosome mosaicism occurs when there is a mixture of chromosomally normal and abnormal cells. True mosaicism may be particularly difficult to detect if present in low frequency. Most cytogenetic analyses are statistically based on an evaluation of a minimum of 15 to 20 cells to reduce the chance to less than 5% probability that true mosaicism will not be detected. A counseling dilemma arises when chromosome mosaicism is observed after chorionic villus sampling or amniocentesis: does the mosaicism detected in the villi or cultured amniotic fluid cells represent the chromosome complement of the fetus? Confirmation of chromosome mosaicism may require additional invasive procedures, such as percutaneous umbilical blood sampling (PUBS), which would place the patient at additional obstetric risk. Even attempts at confirmation of chromosome mosaicism may fail. For example, mosaicism for chromosome 5 present in cultured amniotic fluid cells, followed by normal chromosome analysis of fetal blood obtained by PUBS, still resulted in a child with developmental disabilities because the mosaicism was confined to the skin and was not present in blood cells.17
TYPES OF CHROMOSOME ABERRATIONS AND THEIR CLINICAL SIGNIFICANCE
The normal human chromosome complement represents a balanced genetic constitution: the correct chromosome number and the correct gene content. Deviations from the normal number of chromosomes (i.e. numeric aberrations) or the normal morphologic pattern (i.e. structural rearrangements) represent gains or losses of genetic information from the complement. Genetic imbalances generally disturb the normal developmental processes of fertilization and embryogenesis and result in implantation failure, spontaneous abortion, stillbirth, or a liveborn infant with multiple congenital anomalies. It is possible for a person to carry a chromosome rearrangement but show no clinical effects because there is no accompanying genetic imbalance. Such an individual is called a balanced carrier because the chromosome aberration constitutes a rearrangement of the genetic material into a new chromosome pattern, with no gain or loss of genes. Nevertheless, a balanced carrier is at reproductive risk to transmit the chromosome rearrangement to offspring by gametes that are balanced or unbalanced. A problem arises when prenatal diagnosis reveals the presence of a de novo chromosome rearrangement, because it may not be possible to determine whether or not such a rearrangement is accompanied by a microscopically undetectable gain or loss of genetic material.
Numeric Aberrations
POLYPLOIDY.
The two common forms of polyploidy in human tissues are triploidy, in which each haploid set of 23 chromosomes is present three times (3N = 69) (Fig. 6), and tetraploidy, in which each haploid set is present four times (4N = 92) (Fig. 7). An increased incidence of triploidy and tetraploidy has been reported when oral contraceptive use was stopped within 6 months before conception,18 but this finding has not been confirmed. One percent of all human conceptions are estimated to be triploids. In 66% of the cases, triploidy arises from simultaneous fertilization of an oocyte by two spermatozoa. In the remaining cases, triploidy arises from unreduced oocytes or spermatozoa (i.e. during gametogenesis, the chromosome number is not reduced from 46 to 23).19 Tetraploidy usually arises as a postzygotic event through the failure of cytokinesis (division of the cytoplasm) after chromosome duplication and the incorporation ofdaughter chromosomes into the same cell. Other less likely mechanisms include trispermy and fertilization of a diploid ovum by two haploid spermatozoa or by a diploid spermatozoon.
|

|

A spectrum of clinical effects are associated with polyploidy. Based on studies of preimplantation human embryos, most triploid embryos do not continue to divide and to achieve the blastocyst stage. Triploid embryos that implant frequently abort spontaneously in the first trimester; triploid pregnancies constitute a major etiologic class within the spontaneous abortion population because 15% to 20% of all chromosomally abnormal spontaneous abortuses are triploid.20 A triploid complement has the potential of producing placental hydatidiform degeneration and abnormal fetal development.21,22 Partial hydatidiform mole is found in 80% of triploids.23,24 The roles of the maternal and paternal genome in embryonic development is illustrated in triploidy by comparing the effects of two sets of genes from one parent and one set from the other. For example, when two of the three sets in a triploid are derived from the maternal parent, fetal development is enhanced to the detriment of the placenta. In contrast, when two sets are derived from the paternal parent, placental development is significantly favored over that of fetal development.
A triploid chromosome syndrome has been defined, based on a series of developmental anomalies observed in fetuses aborted electively after prenatal diagnosis and in those rare cases of triploidy that complete pregnancy.22 These anomalies include severe intrauterine growth retardation (IUGR), hydrocephaly, holoprosencephaly, cortical and cerebellar hypoplasia, syndactyly, short halluces, and rocker-bottom feet, and many other physical defects. Chromosome analysis of triploid liveborn infants frequently shows the presence of a normal cell line with 46 chromosomes, which may be responsible for completion of pregnancy but not the prevention of congenital malformations and severe developmental delay.
Tetraploidy frequently is associated with spontaneous abortion in the first trimester, although at least 10 cases of liveborn infants, with chromosome mosaicism in five, have been reported.25 The latter were associated with low birth weight, microcephaly, microphthalmia or anophthalmia, hypertelorism, low-set ears with dysplastic cartilage, micrognathia, arachnodactyly, genital ambiguity, hypotonia, congenial heart disease, and open neural tube defects. Death within 3 months of birth was common, although some newborns with tetraploid mosaicism survived longer. In the course of a chromosome analysis performed on a viable fetus, it is not uncommon to observe complete tetraploidy or diploid/tetraploid mosaicism in chorionic villi and amniotic fluid cells. Almost all of these cases represent cultural artifacts or chromosome changes limited to the sampled tissues and not the fetus proper.
Fetal blood sampling by PUBS may be appropriate for fetuses whose ultrasound evaluations demonstrate abnormal development. The decision to undertake PUBS in all cases of tetraploidy does not have widespread agreement, nor is it considered a standard of care, because PUBS is not without obstetric risks and complications.
Tetraploidy occasionally may occur in solid tumors.26 It does not appear to be specifically related to a particular tumor type, and its relation to cancer is unknown. A series of mutations in genes controlling cell cycle arrest after DNA damage were found to contribute to tumor development by allowing the accumulation of chromosome aberrations and gene mutations leading to cancer.27 These and other biologic processes that impair cytokines may be responsible for the appearance of tetraploidy during carcinogenesis. Alternatively, because polyploidy increases the total gene content, tetraploidy may be one form of cellular response to the development of cancer (i.e. by increasing the total number of genes, a cell may have an advantage over its diploid counterpart).
ANEUPLOIDY.
Aneuploidy, which is the gain or the loss of single, whole chromosomes, constitutes the most important class of chromosome aberrations contributing to human disease. The presence of only one member of a pair of autosomal chromosomes (monosomy) invariably is lethal early in development. Monosomy for the X chromosome (45,X) is the most common chromosome aberration in humans, occurring in up to 5% of all conceptions and 15% to 25% of all first-trimester spontaneous abortions with a chromosome aberration (Fig. 8).28 In 75% to 80% of the cases of monosomy X, nondisjunction of the X and Y chromosomes during spermatogenesis is responsible. Other mechanisms include nondisjunction during oogenesis or after fertilization, with the latter resulting in chromosome mosaicism (e.g. 45,X/46,XX). The clinical features of 45,X are small stature; ovarian dysgenesis, with concomitant amenorrhea and sterility; lymphedema, with puffiness of the dorsum of the hands and feet; shield-like chest, frequently with pectus excavatum; low posterior hairline; cubitus valgus; renal anomalies, particularly horseshoe kidney; redundant skin of the neck or webbed neck; and cardiac and aortic anomalies.28 In cases of chromosome mosaicism, the clinical effects of the monosomic cell line may be mitigated by normal cells.
|
The presence of an additional autosomal chromosome is referred to as trisomy, and it usually results from nondisjunction during gametogenesis. Premature separation of chromosomes with random distribution may actually be the underlying mechanism leading to aneuploid gametes. In trisomic conceptions, 1 of the 22 autosomal chromosomes is present three rather than two times, and the total number of chromosomes in each cell is 47 (Fig. 5). There are several autosomal trisomies (e.g. trisomy 1, trisomy 19) that have not been observed in products of conception or in liveborns, except possibly with a low percentage of mosaicism. There are some common trisomic conditions in abortuses that have never been observed in liveborn infants. For example, trisomy 16 is the most common trisomy among spontaneous abortions, but it is incompatible with life. Autosomal trisomies as a group comprise 15% of all chromosomally abnormal, first-trimester spontaneous abortions. In the case of trisomy 21 (i.e. Down syndrome), the most common trisomy in liveborns, only one in four such conceptions reaches term. Not surprisingly, most trisomic conceptions do not complete term, presumably because of impaired placental function or fetal maldevelopment. The incidence of numeric chromosome aberrations in newborns is 1 in 200, or 0.5%. Other less frequent, but well defined autosomal chromosome syndromes are trisomies 13 and 18. Other trisomies for chromosomes 8, 9, and 22, also producing definable syndromes, generally occur in the mosaic state. The congenital malformations associated with each of these trisomies have been summarized in several compendiums.29,30,31
Chromosome syndromes caused by trisomy share several cardinal features. Autosomal syndromes are characterized by IUGR and postnatal growth retardation, a pattern of dysmorphic features, malformations of several organ systems, and impaired mental development. Congenital malformations consistently associated with trisomy are abnormal facies; low-set, malformed helices; digital anomalies, such as clinodactyly and single palmar creases; and anomalies of the skeletal, cardiac, and genitourinary systems. Neonatal survival is compromised by the presence of these congenital anomalies and shortened life expectancy is characteristic of trisomies 13, 18, and 21. In the latter case, for example, about one third of affected infants die during the first year of life, one half die by age 3 or 4, and the remainder have a reduced life expectancy.32
Females with three X chromosomes (i.e. triplo-X) have clinical features varying from normal to mildly dysmorphic, with and without infertility, to severe retardation.33 Females with more than three X chromosome are uniformly delayed developmentally, with variability in physical features ranging from normal to skeletal and dysmorphic abnormalities, such as microcephaly, radioulnar synostosis, prognathism, and webbed neck.34 XXY males (i.e. Klinefelter syndrome) are characterized by small, firm testes and seminiferous tubule dysgenesis, small penis, gynecomastia, incomplete virilization, variable eunuchoidism, and a tendency to mild mental retardation. In approximately two thirds of the cases of XXY, the two X chromosomes are maternally derived and associated with increased maternal age; when one of the X chromosomes is paternal in origin, maternal age is not increased.35 In individuals with more complex sex chromosome aneuploidy (e.g. 48,XXXY; 49,XXXXY; 49,XXXYY), multiple somatic anomalies and severe developmental retardation are common.35
The XYY chromosome syndrome has drawn great interest to the question of genetic predisposition to criminal behavior since the report of increased prevalence of the YY constitution among tall and mentally retarded males in prisons.36,37,38 Studies during the past 25 years have shown that most YY males are phenotypically normal. Although no anomalous disorders have been observed in YY males originally identified by random newborn screening, a number of physical and personality features have been repeatedly described: excessive height; nodulocystic acne; large deciduous and permanent teeth; subtle neurologic abnormalities, such as intention tremor and incoordination; genital abnormalities and possible increased risk for infertility; increased risk for difficulties with language development; and a questionably increased risk for behavioral disability.39 Prospective studies of YY children have shown no differences in behavioral problems compared with XXY children and controls, but the prevalence of YY males in psychiatric and prison facilities is increased 20 times above that of the incidence of YY males in the newborn population. Early reports characterizing YY males as overly aggressive toward other people have not been confirmed. Reduced intelligence of YY men may increase their chances of being arrested and incarcerated after any antisocial act compared with similar crimes committed by XY men. Because the presence of two Y chromosomes does not impair viability and occurs with an incidence of 1 in 1000 births, the prenatal diagnosis of an XYY fetus is not uncommon, and nondirective genetic counseling is indicated.39
Aneuploidy is an integral part of the biologic processes associated with cancer, particularly those associated with hematologic malignancies, such as leukemia.40 Cytogenetic analyses can contribute to classifying the type of cancer, monitoring the progression of the disease, and determining the effects of treatment. For example, aneuploidy is frequently associated with blast crisis in chronic myelogenous leukemia.
Structural Chromosome Aberrations
Structural changes in the normal banding patterns of chromosomes occur as the result of one of three biologic processes. The most common cause of structural chromosome aberrations is physical breakage involving one or more chromosomes. Two other less common causes of structural chromosome aberrations are unequal crossing over due to mispairing of homologous chromosomes during meiosis and defective DNA synthesis during chromosome replication. When a chromosome breaks, the broken ends are sticky or unstable, and cellular repair mechanisms usually rejoin the two ends promptly (i.e. restitutional healing). In such cases, the occurrence of chromosome breakage would not be detected. However, if more than one chromosome break occurs, the repair mechanisms cannot differentiate between sticky ends and the wrong ends may join (i.e. nonrestitutional healing). Chromosome breakage occurs spontaneously or is induced by exposure to ionizing radiation or mutagenic chemicals. The rate of spontaneous, nonrestitutional chromosome breakage exceeds 1 in 1000 gametes, which is 100 times greater than the spontaneous mutation rate for single gene loci.1
Understanding the nature and behavior of structural chromosome aberrations is of considerable clinical importance. Prospective parents carrying structurally rearranged chromosomes are at significant reproductive risk for genetically unbalanced conceptions. Structural chromosome aberrations also have been the primary means for developing a physical map of the human genome and the first step in the eventual cloning of disease-causing genes.
There are five distinct classes of structural chromosome aberrations. They are translocation, deletion, duplication, inversion, and isochromosome.
TRANSLOCATION.
Translocations are interchromosomal exchanges brought about by breakage and transfer of segments of chromosomes to different locations. There are three basic types of translocations: reciprocal; centric fusion or Robertsonian; and insertional. In a reciprocal translocation, segments distal to breaks in two chromosomes are exchanged. The exchange may involve the long or the short arms of any pair of chromosomes, autosomal or sex (or both), homologous or nonhomologous. Such an exchange usually does not result in any loss of genetic material, and the individual is clinically normal and called a balanced carrier. Nevertheless, a balanced carrier is at significant reproductive risk for producing chromosomally unbalanced gametes that adversely affect conception and embryonic development.
Figure 9 shows a reciprocal exchange between the long arm of chromosome 4 and the long arm of chromosome 13 occurring in a balanced carrier. During gametogenesis, homologous segments of chromosomes normally synapse (i.e. pair together), and in the case of reciprocal translocation between chromosome 4 and 13, this would produce an association of four chromosomes in the form of a cross-shaped quadrivalent. At anaphase of meiosis I, depending on the type of chromosome segregation, 12 different gametes involving chromosomes 4 and 13 are possible, six from 2:2 segregation (two chromosomes into each daughter cell), and six from 3:1 segregation. With the former combination, three types of segregation are possible: alternate, producing one chromosomally normal gamete and the other that is a chromosomally balanced gamete; adjacent I, resulting in gametes that carry duplications and deficiencies for segments of chromosomes 4 and 13; and adjacent II, also producing gametes with deficiencies and duplications of segments of chromosomes 4 and 13. With 3:1 segregation, six other chromosome combinations are possible, but the genetic imbalance causes early pregnancy failure, and in most cases, the pregnancy goes unrecognized. Because centromeres from homologous chromosomes regularly segregate from one another during meiosis I of gametogenesis, 2:2 segregation is more likely, particularly alternate and adjacent I forms. Although in theory gametes with twelve different chromosome complements are possible, in practice the actual number is limited by the pattern of chromosome segregation and their effect on embryonic viability. The actual risk of unbalanced offspring is always lower than the theoretical risk (Table 2).
TABLE 2. Minimal Risk of Chromosomally Unbalanced Conception Detected by Prenatal Diagnosis for Carriers of Balanced Structural Rearrangements
|
| Minimal Risk of |
|
| Chromosomally |
|
| Unbalanced |
Rearrangement | Carrier | Conception (%) |
Centric fusion 13;14 | Father | 1 |
Centric fusion 13;14 | Mother | 1 |
Centric fusion 14;21 | Father | 1 |
Centric fusion 14;21 | Mother | 11 |
Centric fusion 21;22 | Father | 5 |
Centric fusion 21;22 | Mother | 10 |
Centric fusion 21;21 | Father | 100 |
Centric fusion 21;21 | Mother | 100 |
Reciprocal translocation | Father | 12 |
Reciprocal translocation | Mother | 12 |
Pericentric inversion* | Father | 4 |
Pericentric inversion* | Mother | 8 |
* Does not include the common chromosome 9 pericentric inversion
|
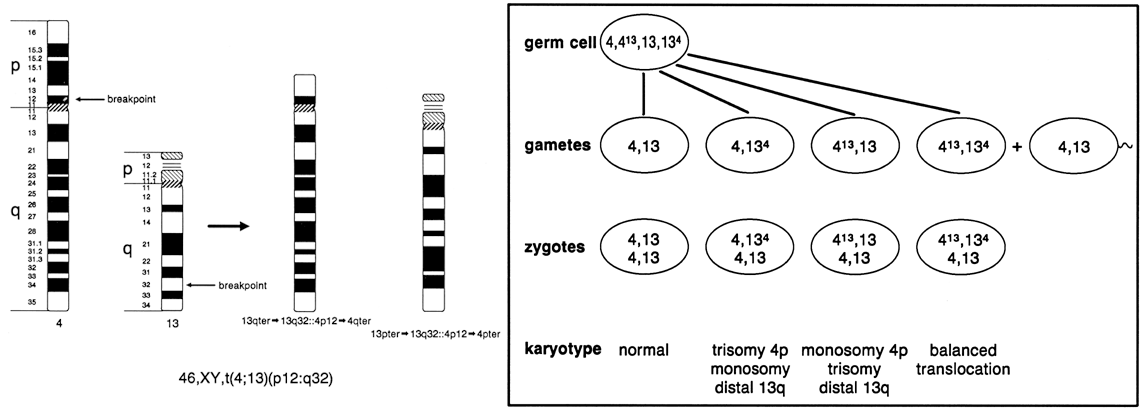
A de novo, apparently balanced translocation detected in the course of prenatal genetic diagnostic studies cannot ensure a normal phenotypic outcome. Balanced translocations have been observed in liveborn infants with congenital malformations and developmental disabilities whose parental chromosomes are normal. A survey of centers performing prenatal diagnosis showed that the risk of an adverse pregnancy outcome with a de novo translocation detected by second-trimester amniocentesis was high, exceeding 9%.41 Although the specific risk of mental retardation and birth defects cannot be predicted with certainty, empirical data indicate that 1 of 10 sporadic, apparently balanced translocations may be associated with loss of normal gene function and abnormal embryonic development. The recurrence risk (i.e. the chance of a second conception with the same chromosome rearrangement) is low, although rare instances of gonadal mosaicism for structural aberrations have been observed. Prenatal diagnostic studies in subsequent pregnancies are warranted in cases of de novo translocations, despite the low risk of recurrence.
Centric fusion, or a Robertsonian translocation, results as a consequence of chromosome breakage at or near the centromeres of two acrocentric chromosomes (13, 14, 15, 21, and 22). The breaks usually occur just above the centromeres, resulting in a single metacentric chromosome with two centromeres (i.e. dicentric) and a satellite fragment with no centromere (i.e. acentric). Any acentric fragment invariably is lost during cell division. Alternatively, Robertsonian translocations may arise in some cases as a consequence of crossing over between homologous segments of two acrocentric chromosomes during meiosis I. In either case, the formation of a Robertsonian translocation is associated with reduction of chromosome number by one, with the diploid number being 2N = 45. The genetic material contained on the short arms of acrocentric chromosomes appears to have no clinical effects. Despite their loss, individuals who carry de novo Robertsonian translocations are phenotypically normal. There is, however, an increased reproductive risk of forming unbalanced gametes during gametogenesis.
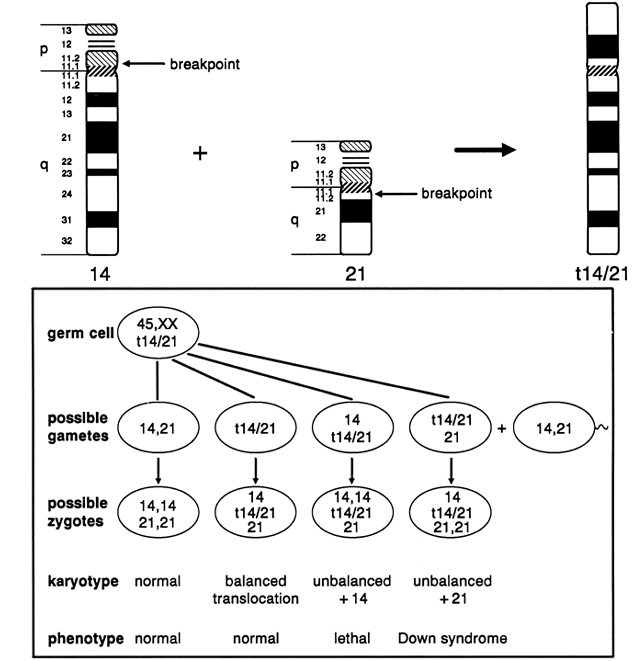
Centric fusion of chromosomes 13 and 14 is the most common structural chromosome aberration in humans, occurring with a frequency of 1 in 1000 individuals.1 The next most common Robertsonian translocation involves chromosome 21 with chromosomes 13, 14, 15, 21, and 22. As shown in Figure 10, a balanced carrier of a 14;21 centric fusion translocation has 45 chromosomes and, although healthy, is at significant reproductive risk. When homologous chromosome segments synapse during meiosis, a trivalent is formed of the two normal chromosomes, 14 and 21, with the 14;21 translocation chromosome. At anaphase I, the three chromosomes can segregate in three ways, producing gametes of six different constitutions. Alternate segregation is associated with one chromosomally normal gamete and one balanced gamete. Adjacent I segregation results in one gamete carrying a duplicated segment of chromosome 14 and one deficient in the same segment; because both are unbalanced, embryonic and/or fetal viability are likely to be compromised, and spontaneous abortion is likely. Adjacent II segregation produces one gamete with duplication of the long arm of chromosome 21 and one gamete with complete deficiency of chromosome 21. Although liveborn infants with only one 21 chromosome (from the other parent) has not been observed, the gamete with duplication of the long arm of 21 results in a conception with Down syndrome. Although six different gametic combinations are theoretically possible, only three types of conceptions are observed: conceptions with a normal complement of 46 chromosomes; a balanced 14;21 translocation carrier with 45 chromosomes and a normal pregnancy outcome expected; and an unbalanced chromosome constitution because of the presence of the 14;21 translocation and two chromosomes 21, resulting in Down syndrome. These three types of conceptions do not occur with equal frequency in the liveborn population, partly because of selection factors favoring the gametes carrying the 14;21 translocation and partly because of the sex of the carrier parent. Empiric data obtained in familial forms of 14;21 translocations indicate that approximately 20% of pregnancies end in spontaneous abortion; as many as 50% of all liveborn infants are translocation carriers like one of the parents. If the maternal parent is the carrier, the risk of a Down syndrome liveborn infant is 11%, whereas if the paternal parent is the carrier, the risk of Down syndrome is less than 5%42 (Table 2).
|
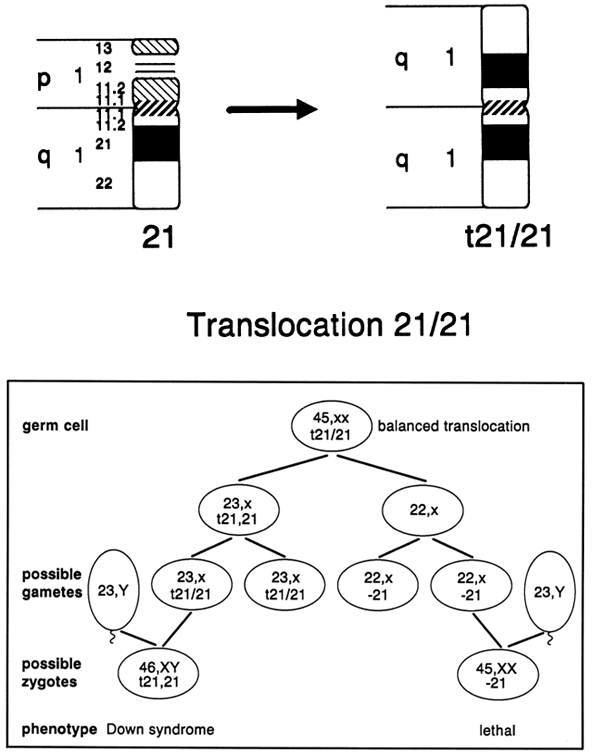
In approximately 4.5% of all newborn cases, Down syndrome occurs as a consequence of an unbalanced Robertsonian translocation involving chromosome 21. Chromosome studies in such newborns, however, show that only one half of the cases are familial. The remaining cases arise de novo in the meiotic cell division producing the gamete that participated in fertilization. Down syndrome as a consequence of a Robertsonian 21;21 translocation presents a unique situation. In approximately 95% of cases, the 21;21 translocation observed in a newborn with Down syndrome has arisen de novo. The remaining cases most likely are caused by gonadal mosaicism, because the origin of a balanced carrier for a 21;21 Robertsonian translocation would require two biologic errors (formation of the translocation in one parental gamete and loss of a normal chromosome 21 in the other parental gamete). If centric fusion of the two 21 chromosomes occurred during gametogenesis, the resulting conception could not be balanced but rather would be affected with Down syndrome because of the normal chromosome contributed by the other parent's gamete. If centric fusion of the two 21 chromosomes occurred postzygotically, presumably there would be cells with 46 chromosomes and cells with 45 chromosomes carrying the translocation, with one unlikely exception: formation occurring before the first cell division of the zygote.
What is unique to carriers of 21;21 translocations is that all of their gametes result in chromosomally unbalanced conceptions (Fig. 11). Approximately one half of the conceptions are affected with Down syndrome, based on the contribution of the 21;21 translocation from one parent and a normal 21 chromosome from the other parent. The chromosome constitution of the remaining conceptions has only one chromosome 21 (monosomy 21), and will not be viable. If all of the cells of a potential parent carry the 21;21 translocation in a balanced state, prenatal diagnosis would be of no value, because all pregnancies are chromosomally unbalanced and developmentally compromised. When Down syndrome occurred secondary to a Robertsonian 21;21 translocation and parental karyotypes were normal, there were no recurrences of Down syndrome.43
|
DELETION.
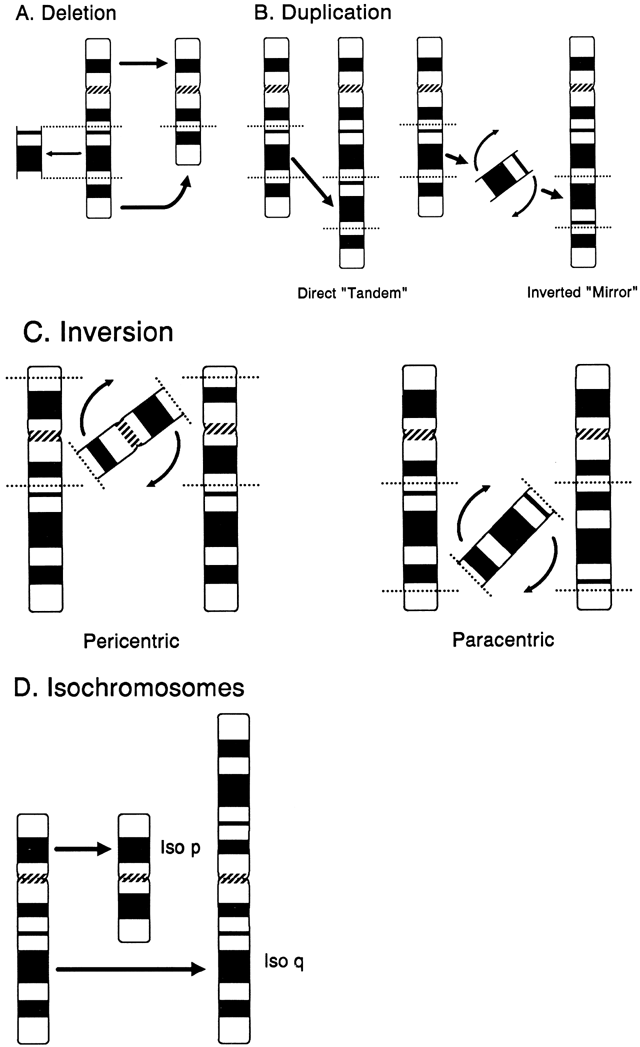
A deletion is the loss of any part of a chromosome. It can occur in several ways. A terminal deletion occurs when a single break in a chromosome produces centric and acentric chromosomes. The latter chromosome, lacking a centromere, is lost in the next cell generation, whereas when the centric chromosome duplicates, the broken ends of the chromatids can fuse together, forming a dicentric chromosome. More commonly, an interstitial deletion arises from the breakage at two points along a chromosome (Fig. 12A). If the chromosome segment between the two sites of breakage does not include the centromere, the acentric fragment so formed will be lost. If breakage occurs in both chromosome arms, the terminal, acentric ends are lost, and the two proximal ends may unite to form a ring chromosome. A ring chromosome with a centromere is usually able to complete mitosis successfully, although if sister-strand crossing over occurs, a ring chromosome that is twice the size of the original ring chromosome, with two centromeres, is formed. Deletion of segments of chromosomes can occur as a consequence of adjacent I and II disjunction involving a parental translocation.
|
The smallest loss of chromosomal material detected microscopically is approximately 400,000 base pairs.1 Conceptions with visible deletions can be monosomic for a large number of genes. With autosomal deletions that are viable, developmental disabilities with mental retardation are to be expected. Specific syndromes associated with deletions include the cri-du-chat syndrome with visible loss of genetic material from the short arm of chromosome 5; the Wolf-Hirschhorn syndrome with deletion in the short arm of chromosome 4; and the Prader-Willi syndrome, with paternally derived deletion of 15q11. Part of a group of syndromes designated as contiguous gene syndromes44 includes conditions that occasionally have been associated with small but cytogenetically detectable deletions or duplications. Included in this group are deletions of 8q24 in Langer-Giedion syndrome,45 deletions of 11p13 associated with aniridia and Wilms tumor,46 deletions of 13q14 associated with retinoblastoma,47 deletions of 17p13 in the Miller-Dieker syndrome,48 and deletions of 22q11 in DiGeorge syndrome.49 When cytogenetic analysis does not show a visible deletion, molecular analysis can demonstrate the loss of specific gene sequences representing submicroscopic deletions of the band or bands previously associated with the specific syndrome. The ability to apply molecular techniques for the cloning of the gene responsible for Duchenne and Becker muscular dystrophies, for example, first required determining its physical location, which was accomplished through deletion mapping.50
DUPLICATION.
Duplication of any segment of a chromosome may arise as the result of unequal crossing over during meiosis or from meiotic events in a parent with a translocation, inversion, or isochromosome (Fig. 12B). In general, duplications as a form of chromosomal imbalance appear to be better tolerated clinically than deletions. At the molecular level, duplications in the form of repeats of specific nucleotide sequences appear to play a primary role in such gene disorders as the fragile-X syndrome, myotonic dystrophy, and the Kennedy syndrome and may be an important feature in many other human genes.51 For example, the fragile-X syndrome is caused by a heritable unstable DNA sequence because of changes in copy number of a trinucleotide repeat, CGG.52
The overall incidence of visible deletions, duplications, or a combination of the two in newborns is estimated at 1 case 1 per 2000.53 For any chromosomal duplication or deletion detected prenatally or postnatally, parental chromosomes must be examined to exclude a balanced structural rearrangement. If parental karyotypes are normal, the recurrence risk is not increased above the general population incidence, with the rare exception of gonadal mosaicism.
INVERSION.
Inversions arise from two chromosome breaks occurring in the same chromosome, followed by a rotation of 180 degrees of the segment between the breaks (Fig. 12C). If both breaks occur in the same arm so that the centromere is not included, the inversion is called paracentric. If the breaks are on either side of the centromere, the inversion is called pericentric. The change in gene order has not been associated with any clinical abnormalities, but inversions place their carriers at reproductive risk as a consequence of their structure and behavior during meiosis. Inversions may interfere with the pairing of homologous chromosomes and thereby reduce crossing over within the inverted segment, but for synapsis to occur, one member must form a loop in the region of the inversion (Fig. 13A). If crossing over occurs within the loop of a paracentric inversion, a dicentric chromatid and an acentric fragment are formed, both of which are unstable and unlikely to result in abnormal offspring. If a single crossover occurs within the loop of a pericentric inversion, the resulting two chromatids have complementing deletions and duplications, and abnormal offspring may result (Fig. 13B).
|
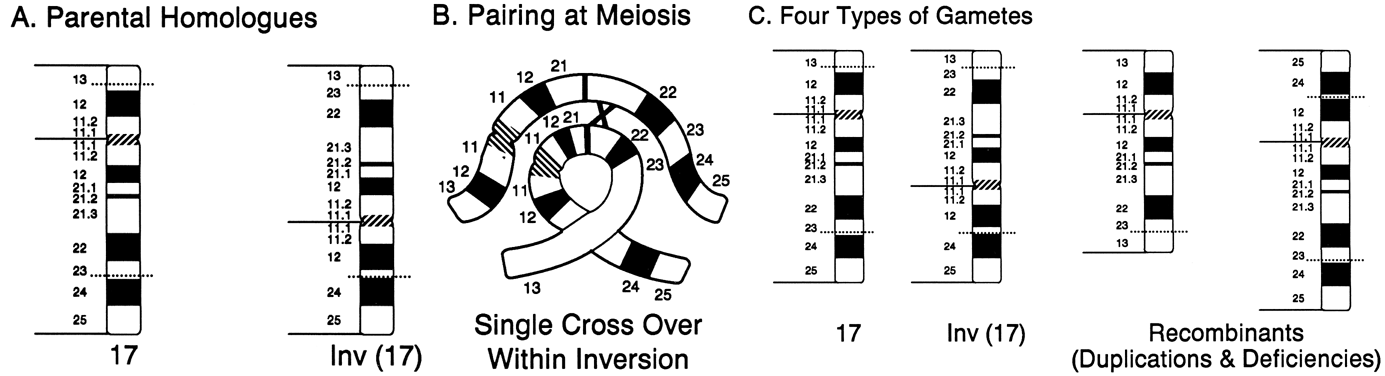
The closer both breakpoints are to the ends of the chromosomes, the greater is the chance that the pregnancy will come to term. Paracentric inversions are not an indication for prenatal diagnosis, because if crossing over does occur within the inversion loop, the degree of genetic imbalance is incompatible with viability in all but the most exceptional instances. Carriers of pericentric inversions, however, are at risk for viable, chromosomally unbalanced offspring, especially when a large inversion is involved. Empirically, this risk is 8% for maternal carriers and 4% for paternal carriers53 (Table 2). One exception is worthy of mention: small pericentric inversions of chromosome 9. These common inversions, which are present in 1% of the general population, have not been associated with abnormal offspring as a consequence of crossing over within the inverted segment.
ISOCHROMOSOME.
An isochromosome is a structurally altered chromosome in which there is deletion of one entire arm and complete duplication of the other arm (Fig. 12D). This type of chromosomal aberration most commonly arises by misdivision of the centromere in the transverse direction. Another mechanism involves the simultaneous breakage of both chromatids (i.e. isochromatid breakage), followed by fusion of the centromeric portions and resulting in a dicentric chromosome. When the isochromosome is dicentric, one centromere apparently becomes nonfunctional so that the isochromosome routinely segregates normally during cell division. Isochromosomes for the long arms of the X and Y chromosomes have been observed in liveborn infants, whereas for other chromosomes, an isochromosome usually leads to an early spontaneous abortion, with two rare exceptions—isochromosomes for the short arms of chromosomes 9 and 12.1
Special Characteristics of the X and Y Chromosomes
The X and Y chromosomes exhibit certain unique biologic properties that are clinically relevant to the practice of obstetrics and gynecology.
X INACTIVATION.
According to the Lyon hypothesis, in somatic cells, one of the two X chromosomes becomes inactivated early in embryonic life. The inactivation is random; the maternal or the paternal X chromosome may be inactivated. X inactivation is virtually complete (i.e. almost all of the genes on the X chromosome are inactivated). X chromosome inactivation is permanent and clonally propagated, and if a maternally derived X chromosome is inactivated in a parental cell, an active paternally derived X chromosome is expressed in all of the daughter cells, whereas the maternally derived X remains inactive.54 The Lyon hypothesis explains the phenomenon of dosage compensation in females wherein the amount of gene product of X-coded genes is the same in females with two X chromosomes as in males with one X chromosome.
Several important cytogenetic observations contributed to the development of the Lyon hypothesis. First and foremost was cytologic evidence of the existence of sexual dimorphism: only the nuclei of female cells contain a dark-staining chromatin body approximately 1 μm in diameter in juxtaposition to the nuclear membrane (the X-chromatin mass, or Barr body). Subsequent studies in humans with more than two X chromosomes showed that the number of X-chromatin bodies was equal to the number of X chromosomes minus 1. Whereas 46,XY males exhibit no X-chromatin mass in their cells during interphase and 46,XX females exhibit one X-chromatin mass, 47,XXX and 48,XXXY individuals have two X-chromatin bodies in their cells. Studies also showed that during prophase, one of the two X chromosomes appeared to be heteropyknotic (i.e. dense and dark-staining), which suggests gene inactivity. In vitro studies monitoring the incorporation of bromodeoxyuridine (BUdr) into DNA indicated differential replication of the two X chromosomes: the inactive X chromosome was the last chromosome to replicate its DNA during the S (i.e. synthesis) phase of mitosis and hence the term, “late replicating, was applied to the inactive X chromosome.55 In a patient with 49,XXXXX aneuploidy, the cells exhibit the presence of four X-chromatin bodies, four heteropyknotic X chromosomes during prophase, and four late-replicating X chromosomes in the S phase of mitosis, all of which provide cytologic evidence of genetically inactive X chromosomes.
The Lyon hypothesis holds the key to several medically significant problems. It provides the biologic basis for the greater variation in clinical manifestations of X-linked genes and diseases in heterozygous females, compared with hemizygous males. In the same manner, it explains past difficulties in using biochemical techniques to identify carrier females of X-linked gene mutations and why this approach has been replaced, wherever possible, by X-linked DNA markers. Lyonization has provided insight into sex determination in humans because the gene action of the Y chromosome is basically interacting with only one functional X chromosome, despite the presence of two or more X chromosomes.
A paradox remains, however, in that individuals with more than two X chromosomes may not be developmentally normal, and the more X chromosomes a patient has, the greater the likelihood of mental retardation. X chromosome inactivation during human embryonic development is believed to occur during the blastocyst stage of implantation. An interesting observation whose biologic significance has yet to be determined is the fact that the paternal X chromosome is inactivated preferentially in placental tissue and other extraembryonic membranes. X chromosome inactivation also has been used as a marker in studies of differentiation and malignancy. For example, in a study of uterine fibroids in women who were heterozygous for glucose-6-phosphate dehydrogenase alleles, A and B, each fibroid tumor expressed only the A or B type, but never both. This finding indicated that each tumor arose from the clonal propagation of a single cell rather than a multicentric origin.53
The existence of a specific site on the X chromosome responsible for the process of X chromosome inactivation was first inferred from studies of X autosome translocations in humans and from mapping of a murine locus, Xce, which influences preferential inactivation of an X chromosome in a cis-acting manner.56 The location of the X inactivation center (XIC) in humans has been determined to be located at Xq13, although the DNA sequence itself remains unresolved. A transcript that is exclusively transcribed from the inactive X chromosome has been identified, the X-inactive-specific transcript (XIST).57 A close relation exists between inactivation of the X chromosome and expression of XIST.58,59,60 XIST is expressed during the period in male germ cell development in which the single X chromosome is transiently inactivated and XIST expression ceases at a time in female germ cell development when the inactive X chromosome becomes reactivated. A number of questions remain.61 Does XIST expression lead to inactivation of the X chromosome, or does XIST expression occur because the X chromosome has become inactivated? How does the cell maintain one and only one active X chromosome? If XIST has a functional role in X inactivation, how is this accomplished? Answers to these questions will further understanding of the nature of X chromosome disorders and possibly lead to methods of amelioration.
Y CHROMOSOME.
In humans, the Y chromosome is responsible for normal male development. Phenotypic-karyotypic correlations in males with structural aberrations of the Y chromosome first contributed to localizing genes affecting stature and testicular development. The entire Y chromosome has been cloned using molecular techniques,62 setting the stage for the complete gene mapping of the Y chromosome.63 The presence of an XY karyotype or a structurally modified Y chromosome in a patient with a female habitus can present difficult clinical challenges. In the case of 45,X/46,XY chromosome mosaicism, the phenotype ranges from almost normal males with cryptorchidism or penile hypospadias to females indistinguishable from those with 45,X (i.e. gonadal dysgenesis). A diagnosis of 45,X/46,XY is likely if the patient has a unilateral streak gonad and a contralateral testis, or bilateral dysgenetic testes and mullerian derivatives.64 Approximately 5% of patients with gonadal dysgenesis and unambiguous external genitalia have cells containing a Y chromosome in addition to cells with 45,X. The dysgenetic gonads may undergo transformation into a gonadoblastoma or dysgerminoma. Most 45,X/46,XY persons have ambiguous genitalia and gonads consisting of a unilateral streak one side and a contralateral dysgenetic testis (i.e. mixed gonadal dysgenesis). The risk of neoplastic transformation ranges between 15% and 20%. Breast enlargement in a 45,X/46,XY individual with ambiguous external genitalia is clinical evidence of a gonadoblastoma or dysgerminoma.64 A paradox exists in that, when 45,X/46,XX is detected prenatally, the fetus invariably has normal male external genitalia, with the only abnormalities being hypospadias or unilateral cryptorchidism.65
Subjects with the XY form of pure gonadal dysgenesis (i.e. Swyer syndrome) have bilateral streak gonads, female external genitalia, müllerian derivatives (i.e. uterus and fallopian tubes), and no somatic abnormalities.66 The familial form of Swyer syndrome is inherited as an X-linked recessive or a male-limited autosomal dominant gene. In many sporadic forms and some familial cases, microdeletions of the Y short arm containing the testisdetermining factor (TDF) can be responsible for Swyer syndrome.67 In addition to the effects of hypogonadism and infertility, the XY form of pure gonadal dysgenesis is associated with a 30% lifetime risk of gonadal neoplasia.
Fragile Sites
Fragile sites describe the cytologic observation that segments of chromosomal material appear as nonstaining gaps of variable width, usually involving both chromatids. A fragile site is always observed at the same point on the chromosome in cells studied from a subject. It is inherited in a mendelian codominant fashion. Molecular studies are beginning to elucidate the structural and functional natures of fragile sites. A fragile site may represent a specific segment of a chromosome that does not undergo normal condensation in mitosis. The loci of several oncogenes and the sites of chromosomal breakage that lead to structural rearrangements correlate with sites of fragility. The most clinically relevant fragile site is that of the X chromosome at band q27, which accounts for one third of all mentally retarded males and females (Fig. 14). The effect on the human population is significant, because the frequency of the fragile-X syndrome is approximately 1 in 1500.
|
Molecular studies have resulted in identification of a gene, FMR1, containing a trinucleotide repeat sequence, CGG, coincident with the fragile-X syndrome.52,68 In fragile-X syndrome, a CGG repeat in the 5' untranslated region of the FMR-1 transcript normally is polymorphic, displaying alleles ranging from 6 to 60 repeats (mean, 29). This repeat is unstable in fragile-X families with repeat lengths varying from 60 to approximately 200 triplets in nonpenetrant carrier females and massively expanded well beyond 200 repeats, often up to 1000 triplets, in penetrant males. About one half of carrier females with more than 200 copies are mentally impaired. Expansion beyond 200 repeats is accompanied by methylation of the repeat region, which results in transcriptional silencing of the gene and the clinical expression of the fragile-X syndrome. Increases in the length of the CGG repeat in fragile-X carrier females is correlated with proportional increases in risk to full expansions and penetrant offspring.69 This phenomenon is similar to genetic anticipation, in which increasing clinical involvement and decreasing age of onset occur in successive generations.
CAUSES OF CHROMOSOME ABERRATIONS
The origins of the different chromosome aberrations are well understood: polyploidy from failed cytokinesis, aneuploidy from nondisjunction or premature separation of chromosomes, and structural rearrangements from chromosome breakage. These descriptive terms, however, do not indicate underlying causes of chromosome aberrations. A host of intrinsic and extrinsic factors produce chromosome aberrations in tissue culture cells and experimental animals. Polyploidy has been produced by temperature shock and by a number of chemical agents that interfere with cytokinesis or spindle fiber formation. Aneuploidy has been attributed to specific genes influencing meiotic and mitotic segregation, to advancing maternal age, and to the presence of structural rearrangements. Structural chromosome aberrations have been induced by ionizing radiation, viruses, and exposure to a variety of chemical agents.70 Nevertheless, evidence that any of these agents is clinically relevant in humans is inconclusive, with exception of the positive association of advanced maternal age and the occurrence of nondisjunction.71
Aneuploidy in humans has also been attributed to prior maternal x-ray exposure, parental metabolic impairment and dietary deficiencies, and delayed fertilization.72 These and other factors have yet to be established as the underlying cause of X chromosome monosomy, Y disomy, or autosomal trisomy. Similarly, agents capable of causing breakage of human chromosomes in experimental settings have yet to be positively identified as the cause of pregnancy wastage or birth defects. Chromosome aberrations have been observed in individuals exposed to various clastogenic agents because of occupational accidents, a need for health care, or their residence.73 The clinical relevance of these exposures toward reproductive outcomes such as spontaneous abortion remains difficult to interpret, generally or in any specific instance. Part of the problem is that exposure effects are difficult to establish and quantitate: chromosome aberrations rarely occur; unstable chromosome rearrangements that are formed, such as acrocentrics, dicentrics, and rings, are likely to be lethal and selectively eliminated in the gametic or preimplantation stages; and obtaining an adequate sample size large enough to permit definitive conclusions is not likely.73
Although there are many causes of chromosome aberrations, the cause of clinically significant chromosome aberrations remains unknown. It has not been shown conclusively that exposure of prospective parents to agents that affect cell division or chromosome breakage causes chromosome aberrations in their offspring.
FREQUENCY OF CHROMOSOME ABERRATIONS
Chromosome aberrations occur at each defined stage of biologic development. Chromosomes of human spermatozoa can be visualized and analyzed if the sperm fertilize hamster oocytes denuded of their zona pellucida. These studies have shown that normal males with a negative family history exhibited chromosome aberrations in 10.8% of their ejaculates, with a range of 0% to 24%.74 Oocytes of varying quality have been obtained in conjunction with programs of assisted reproduction using in vitro fertilization. In one multicenter study,2 karyotypic analysis of oocytes that were acceptable for fertilization and embryo transfer showed that 26% contained chromosome aberrations. Oocytes that were rejected on the grounds of poor morphologic features or because they failed to be fertilized contained an even higher percentage of chromosomal abnormalities, up to 89%. Most were numeric in type.75 The multicenter study also found that 29% (range, 21% to 71%) of preimplantation embryos carried a chromosomal defect.2,76 It was estimated that one half of these embryos fail to implant and that 96% of these chromosomally abnormal embryos that implant do not complete the first trimester. Aneuploidy was estimated to be present in 0.6% of second- and third-trimester fetuses.2
Approximately 50% to 70% of all spontaneous abortions are associated with chromosome aberrations: in the first trimester, 60%, and between 12 and 20 weeks, 20%.77 For women of advanced maternal age ( 33 years) undergoing first-trimester chorionic villus sampling (CVS), the overall frequency of chromosome aberrations was 2.5%. The procedure was limited to women with viable pregnancies whose growth parameters were compatible with gestational age. For the same women undergoing second-trimester amniocentesis, 1.5% had karyotypically abnormal pregnancies.78 In stillborns and neonatal deaths, the frequency of chromosome aberrations ranged from 5% to 10% and frequently involved trisomies for chromosomes 13, 18, and 21.79 Chromosome analysis on stillbirths and in cases of neonatal deaths associated with multiple malformations has considerable clinical importance because it may identify a familial structural rearrangement. Clinically significant chromosome aberrations occur in 0.65% of all newborns, and an additional 0.2% are born with balanced structural chromosome rearrangements that place them at markedly increased reproductive risk later in life.
33 years) undergoing first-trimester chorionic villus sampling (CVS), the overall frequency of chromosome aberrations was 2.5%. The procedure was limited to women with viable pregnancies whose growth parameters were compatible with gestational age. For the same women undergoing second-trimester amniocentesis, 1.5% had karyotypically abnormal pregnancies.78 In stillborns and neonatal deaths, the frequency of chromosome aberrations ranged from 5% to 10% and frequently involved trisomies for chromosomes 13, 18, and 21.79 Chromosome analysis on stillbirths and in cases of neonatal deaths associated with multiple malformations has considerable clinical importance because it may identify a familial structural rearrangement. Clinically significant chromosome aberrations occur in 0.65% of all newborns, and an additional 0.2% are born with balanced structural chromosome rearrangements that place them at markedly increased reproductive risk later in life.
Estimates of chromosome aberrations among persons with reproductive or other medical problems vary according to the nature of the problem. Among couples experiencing three or more spontaneous abortions, the overall incidence is 1 per 14 patients (reports range from 1 in 100 to 1 in 7).80 Ten percent of subfertile males with low sperm counts have demonstrable chromosome aberrations.81 Although accurate estimates are not available, institutions for the mentally retarded clearly have a higher proportion of patients with trisomy 21 and fragile-X syndrome than the general population, whereas penal institutions are likely to have a relatively high frequency of inmates with 47,XYY and 47,XXY associated with behavioral disorders.36,37,38
CLINICAL CONDITIONS APPROPRIATE FOR CYTOGENETIC ANALYSIS
An internationally recognized standard of care is to offer prenatal diagnostic testing, CVS or amniocentesis, to women of advanced maternal age, to prospective parents who have had aneuploid offspring, and when one parent is a balanced carrier of a chromosome rearrangement. Second-trimester amniocentesis is also recommended when ultrasound evaluation detects fetal anomalies such as hydrocephalus or renal maldevelopment, or when multiple marker screening (i.e. maternal serum α-fetoprotein, unconjugated estriol, and human chorionic gonadotropin) indicates a risk of trisomy 21 greater than 1 in 250 to 290 at the time of amniocentesis.
Persons with the following clinical conditions warrant chromosomal analysis of their peripheral blood lymphocytes:
Reproductive problems, such as infertility and repeated pregnancy loss, for which other causes have been excluded
Birth of a child with multiple congenital malformations or developmental disabilities
Detection of fetal anomalies with ultrasound
Abnormal sexual development
Mental retardation or physical anomalies of unknown origin
Family history of developmental disabilities, recurrent pregnancy losses (including stillbirth), and mental retardation
For patients with certain forms of malignancy, such as chronic myelogenous leukemia, chromosome analysis of bone marrow cells is indicated. Establishment of a chromosomal cause for any of these clinical conditions can aid in diagnosis, management, and prognosis. In certain instances, clinical management may be altered depending on the results of the cytogenetic analysis. For example, detection of a balanced chromosome rearrangement may be useful in counseling about the possibility of recurrent spontaneous abortion and conceptions with developmental anomalies, the risk and benefits of antenatal diagnosis, and the existence of reproductive alternatives, such as tubal ligation and adoption.
Intrauterine Growth Retardation
Genetic causes of IUGR include all of the chromosomal aneuploidies (e.g. trisomy 21, monosomy X) and syndromes assumed to be single-gene mutations, particularly for any stillbirth or perinatal death associated with IUGR. There are two general types of IUGR: symmetric, in which case the fetus is small in all measurements, and asymmetric, in which the head is relatively spared in relationship to the corpus growth retardation. Symmetric IUGR is thought to have a constitutional cause (e.g. autosomal trisomy), whereas asymmetric IUGR is thought to be nutritional in cause (e.g. uteroplacental insufficiency).
Another possible cause of IUGR is that of confined placental mosaicism (CPM).82,83 True chromosomal mosaicism involves the presence of two or more cell lines having different chromosome complements in the same person. In CPM, the fetus presumably has a normal diploid genotype, whereas the placenta is mosaic for a chromosome aberration, usually trisomy for a specific chromosome. CPM most likely originates from the postzygotic loss of one of the three chromosomes in the embryonic progenitors. CPM has been detected for a variety of chromosomes, particularly trisomies 2, 7, 9, 12, 15, 16, and 22. The overall incidence of CPM in all gestations has been reported to be 1% to 2%, but the rate of CPM is higher in spontaneous abortuses and in cases of IUGR.83,84 Approximately 20% of spontaneous abortions and 6% of fetuses with IUGR have CPM. Approximately 50% of fetuses with CPM continue to term and are viable, although 16% to 21% of them have perinatal complications.
Preliminary evidence suggests that the normal chromosome complement in CPM may be beneficial to the development of the fetus. Many trisomies are lethal if they are not mosaic, so that the confinement of the extra chromosome to the placenta may contribute to maintaining viability. Trisomy 13 and trisomy 18 fetuses, but not trisomy 21 fetuses, were more likely to reach term and be born live if CPM for a diploid line confined to the cytotrophoblast was present.85 The conclusion was that the normal diploid portion of the placenta supported development to term and that, without this CPM, the ability to progress to term was greatly reduced.
Most fetuses with CPM do not have abnormal clinical outcomes and can be completely normal at birth and throughout life. Some cases that have had adverse outcomes, including cases of IUGR and spontaneous abortions, have apparently resulted from the inheritance of both members of a pair of chromosomes from one parent. Uniparental disomy, as this arrangement is called, may have as its origin CPM.86,87 In the case of a trisomic zygote, for example, subsequent chromosome segregation may lead to unequal distribution, causing extraembryonic tissues to contain two maternal and one paternal copy, whereas the embryo proper contains two maternal copies. This maldistribution in the embryo is likely to result in IUGR and severe perinatal morbidity or mortality. The clinical effects also depended on which chromosome, maternal or paternal, was isodisomic. In were instances, for example, IUGR and severe perinatal morbidity or mortality were associated with maternal isodisomy, whereas cases with paternal disomy were normal. In some cases, the placental trisomy, caused by an extra copy of the maternal chromosomes (two maternal copies and one paternal copy) with a normal biparental dose of the two chromosomes in the fetus, was not associated with increased morbidity or IUGR. These findings show that the phenotype must be associated with the embryonic effect, not the placental effect. The paternal chromosome is required for normal, or non-IUGR, development.88,89,90
The observation of these phenomena in human and other mammalian systems has led to the isolation of chromosomal regions and, in some cases, specific genes that are imprinted such that only one parent's genes are expressed and the other parent's gene are silent. There are an increasing number of human syndromes in which imprinted genes play an important role. Beckwith-Wiedemann syndrome, for example, is a rare disorder that appears to be inherited in a manner that strongly supports the concept of an imprinted gene.88 Affected individuals carry paternal disomy for the specific chromosomal region, 11p15. The syndrome is characterized by generalized overgrowth of fetal and placental tissues, omphalocele, protruding tongue, hydropic placenta, massive fetal adrenal cortical cytomegaly, and a unique predisposition to developing tumors, such as adrenal cortical adenocarcinomas and Wilms tumors. The gene has been localized to 11p in a region whose homologous murine region includes the maternally imprinted gene for insulin-like growth factor type 2 (IGF2) (i.e., the maternal copy is transcriptionally silent, and only the paternal copy is active). IGF2 may be the candidate Beckwith-Wiedemann syndrome gene.91
Hydatidiform Mole Versus Teratoma
Long-standing evidence shows that both parental haploid genomes were essential for normal human development. When only the paternal genome is present (i.e. diploid karyotype of only paternal DNA), the gestation progresses as a complete hydatidiform mole. In the opposite cases, in which only a maternal genome is present, a teratoma results.92 In humans, the paternal genome apparently is required for normal extraembryonic development, whereas the maternal genome has a more important role in embryogenesis. Nevertheless, neither genome can function normally without the other. Similar outcomes occur in the case of triploid conceptions. Triploidy derived from an extra haploid paternal genome results in a partial hydatidiform mole with a characteristic trophoblast hyperplasia and a small, but distinct risk for the development of persistent trophoblastic disease. The resulting phenotype is different, however, when the triploid gestation carries an extra haploid maternal genome. The placenta is small and sclerotic and does not have trophoblast hyperplasia. These fetuses may develop to term, but they have a characteristic phenotype associated with multiple structural malformations that are lethal.
Fetal Abnormalities
Fetal karyotyping may be indicated if the following fetal abnormalities are detected: cystic hygromas, nonimmune hydrops fetalis, idiopathic polyhydramnios, choroid plexus cysts, pelviectasis, and macerated stillbirth. In the case of cystic hygromas, besides monosomy X, in which they may occur in 50% of cases, several other chromosomal disorders, such as trisomies 13, 18, and 21, have been reported in association with this developmental anomaly.93 Chromosome aberrations have been present in 30% to 50% of the fetuses when cystic hygromas was diagnosed in the first trimester of pregnancy.93,94 Among the many causes of nonimmune hydrops fetalis, approximately 10% of cases are caused by chromosome aneuploidy.95 The incidence of aneuploidy detected in patients with idiopathic polyhydramnios (3.2%) was much higher than the reported incidence of chromosome aberrations in liveborn infants (0.59%);96 However, because testing was performed at the beginning of the third trimester, the findings may have limited clinical value. After the detection of choroid plexus cysts or pyelectasis, the risk of a chromosome aberration may be as high as 3%, which exceeds the risk for a 35-year-old woman. Second-trimester amniocentesis appears warranted.97,98 In the case of a macerated stillbirth, the extraembryonic tissues, including the placenta, may have retained their viability and offer excellent prospects for karyotyping, despite the fetal demise.
Germ Cell Neoplasia
The field of cytogenetics has contributed significantly to an understanding of the origin and nature of various cancers. This relationship, in the context of obstetrics and gynecology, warrants discussion. Aspects of germ cell tumor cytogenetics are relevant to an understanding of their natural history and prognosis. First, the extent to which the tumor stem cell has progressed through meiotic development before neoplastic transformation occurs correlates with histologic subtype and natural history. Second, many malignant lesions have a marker chromosome, an isochromosome for the short arm of chromosome 12 [i(12p)], which is specific to germ cell tumors and is of diagnostic importance.99 Third, the risk of tumor formation in individuals with dysgenetic gonads correlates highly with the presence of a Y chromosome. Prophylactic gonadectomy is indicated for these patients because of a greatly increased risk of the development of some type of germ cell tumor, especially gonadoblastoma.68 In the case of ovarian mature teratomas, the stage of origin has been determined by using centromeric markers, such as Q banding. The findings indicated that most of these tumors arise from an oocyte that has completed the first meiotic division in a manner analogous to parthenogenesis.100 Specific, nonrandom cytogenetic abnormalities may be useful as diagnostic tools, because they provide insight into the early events of neoplastic transformation. For example, the isochromosome involving the short arm of chromosome 12 is present in 70% to 90% of testicular germ cell tumors, and it is frequently present in ovarian dysgerminomas and yolk sac tumors.99
INTERPRETATION OF LABORATORY REPORTS
Laboratory interpretation of a chromosome analysis is based on the International System for Human Cytogenetic Nomenclature (ISCN) that has been established through periodic, worldwide conferences of cytogeneticists.101 At a minimum, the laboratory report should describe the chromosome number, the sex chromosome complement, and the presence of any structural chromosome rearrangement. Artifact chromosome changes routinely arise during tissue culture, and not all cells necessarily have 46 chromosomes.
Determining the significance of any chromosome change relies in part on established criteria and in part on the technical expertise and clinical experiences of the cytogenetic personnel. For example, there are established rules for distinguishing true mosaicism from pseudomosaicism. True mosaicism, the presence of more than one type of chromosome complement, is distinguished operationally from in vitro artifacts by finding two or more cells of exactly the same aberration in at least two independently initiated colonies. If true mosaicism is present, physical or developmental anomalies are highly likely, although there are numerous examples where a low frequency of the abnormal cell line is not associated with any phenotypic abnormalities. Chromosome mosaicism detected antenatally presents significant interpretive problems for health professionals and prospective parents. In the U.K. collaborative study on the diagnostic accuracy of CVS, mosaic cytogenetic abnormalities were confirmed by amniocentesis or in fetal tissues in only 23% of cases. Clinical follow-up after detection of mosaicism in amniotic fluid cultures indicated a 5% to 10% risk of anomalies.102 A detailed ultrasound evaluation performed by an experienced ultrasonographer thoroughly trained in embryonic and fetal development and a chromosome analysis of fetal blood cells by PUBS may prove beneficial and reassuring.
MOLECULAR CYTOGENETICS
Recombinant DNA technology has provided methods for generating highly specific DNA and RNA molecular probes that recognize specific genetic sequences with high reliability. Similar advances in molecular technology have also provided methods for labeling these probes and identifying their binding sites when hybridized directly to chromosome preparations. Combining molecular genetics with traditional cytogenetics has established a new era in clinical genetics, that of molecular cytogenetics. The advantages of this approach are based on the rapidity of diagnosis, the highly specific and reliable recognition of the presence or absence of a particular DNA sequence for which the probe has nucleotide sequence complementarity, the simplicity of the methodology, the stability of the signal, and the simple microscopy required for analysis. Multiple gene sites can also be screened through use of sets of multiple probes. Contiguously related probe sets can evaluate larger DNA segments, such as a band, several bands or even entire chromosomes. This technology is likely to make significant changes in the role of cytogenetics in the practice of obstetrics and gynecology over the next decade.103,104
Molecular cytogenetic analysis involves simple DNA denaturation followed by probe hybridization and label recognition. These studies can be performed on the chromosomes of peripheral lymphocytes, cancer cells, amniotic fluid cells and chorionic villi. It also is possible to perform this type of cytogenetic analysis on nondividing, or interphase, cells, bypassing the need for tissue culturing and allowing analysis of biologic tissues heretofore extremely difficult because of their low mitotic indices (e.g. solid tumors). This approach is in stark contrast to conventional cytogenetic techniques, which can involve extensive tissue culturing to generate sufficient high-quality metaphase spreads to obtain suitable karyotypes.
Originally, detection of a specific nucleic acid sequence in cytologic preparations involved the use of DNA or RNA probes labeled with radioactive isotopes (32P or 35S), which were hybridized to metaphase chromosome spreads. A homologous site of the chromosomal DNA sequence was indicated by location of silver grains along the chromatids after autoradiography and staining. This approach, although highly specific and sensitive, had considerable drawbacks. The method was tedious, technically demanding, and time consuming, with exposures to a radioactive nucleic acid probe requiring weeks to months. The use of radioactive probes required isolated laboratories and stringent safety precautions in their use and disposal.
The next technical improvement was the introduction of nonradioactive fluorescence methods in combination with the procedure of in situ hybridization of a nucleic acid probe to target DNA.105 Fluorescent in situ hybridization (FISH) is a technique whereby a nucleic acid probe is typically labeled with nucleotides that are modified with biotin or digoxigenin. Hybridization of the probe to the specimen required several hours to overnight. Visualization of hybridized probes for fluorescence microscopy is achieved using fluorochromes such as fluorescein or Texas red, which are conjugated to avidin for detection of biotin or anti-DIG for detection of digoxigenin.106 This method of indirect labeling of FISH probes now has been improved by directly labeling the FISH probes. Directly labeled FISH probes are reliable, simple to use, and permit a rapid completion of cytogenetic analysis within 1 to 2 hours.
There are three general types of probes that address different applications of the FISH technology. There are chromosome-specific repetitive sequence probes for centromeres, which are suited to detect numeric chromosome aberrations in interphase cells (Fig. 15A); whole chromosome probes which can screen for different chromosomes simultaneously (Fig. 15B); and probes for specific chromosome regions, with a theoretical limit of resolution of a single base pair (Fig. 15C). FISH has been applied to prenatal genetic testing for autosomal trisomies using interphase cells,107 for interpreting chromosomal rearrangements in normal and neoplastic tissues, and for evaluating specimens not suitable for conventional cytogenetic analysis. Specific applications include the identification of marker chromosomes, determination of chromosome mosaicism, rapid assessment of polyploidy, and exclusion of a chromosome disorder in autolyzed tissue from a fetus with structural anomalies.103,104 Analysis of cancer cells by FISH technology showed many specific deletions at different loci and referred to as loss of heterozygosity. Several syndromes have also been associated with deletions detected by molecular cytogenetic techniques, including Kallmann syndrome (i.e. hypogonadotropic hypogonadism and anosmia), congenital ichthyosis due to steroid sulfatase deficiency seen with a deletion involving chromosome band Xp22.3, Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosa, and the McLeod erythrocyte abnormality, seen in some patients with a deletion involving Xp21.104
|
The most direct method of physical gene mapping, the identification of the chromosome locus of a human gene, is accomplished by in situ hybridization. FISH has been used to detect single copy genes in interphase cells108 and unique sequences in G-banded and R-banded chromosomes.109 Commercial availability of FISH probes has greatly expanded the use of in situ hybridization in basic research on chromosome structure and function and in the clinical practice of reproductive and medical genetics.
Until recently, molecular probes were applied to limited targets, such as specific gene sequences, centromeres, and individual chromosomes, through the use of chromosome painting. Because analyses based on molecular cytogenetic techniques were probe specific, the use of multiple chromosome probes did not permit the entire chromosome complement to be evaluated. A chromosome aberration that does not involve the segments or chromosomes complimentary to the applied probes goes undetected. Although molecular cytogenetic techniques proved to be powerful tools for screening certain chromosome disorders, such methods did not obviate the necessity of completing a traditional chromosome analysis.104 Two approaches have now addressed this issue: comparative genome hybridization (CGH)110 and multitargeted FISH.111
CGH is a molecular cytogenetic technique capable of detecting deletions and duplications in the entire chromosome complement by FISH. In CGH, DNA from the cells of a patient are labeled with a fluorescent moiety of one color (e.g. green fluorescence) and mixed with DNA from a reference source labeled with a fluorescent moiety of a different color (e.g. red fluorescence). Equal amounts of probes are mixed and then hybridized to the chromosomes in cells of a normal male. Green to red fluorescence intensity ratio profiles are calculated for each of the 46 chromosomes in a set 10 to 15 cells in the metaphase stage. Based on green-to-red ratios, trisomies, monosomies, and partial chromosome gains or losses can be identified (Fig. 16). CGH screens the entire genome for unbalanced chromosome aberrations in a single experiment, and is a rapid and accurate technique for the detection of extra or missing chromosome material without requiring actively dividing cells. The CGH technique is particularly suitable in cancer genetics, primarily as a research tool but eventually may be useful for clinical cytogenetics as well.
|

Multitarget FISH provides the means to detect multiple genetic targets simultaneously within nuclei or metaphase spreads (Fig. 17). The technique combines probes for a number of targets into a single FISH hybridization. By simultaneously painting each of the 24 chromosomes in a different color, it is possible to detect several chromosome aberrations within a single cell. Although considered experimental, this approach has proven useful in cytogenetic analyses of neoplastic tissues in cases where multiple chromosome aberrations are present.
|
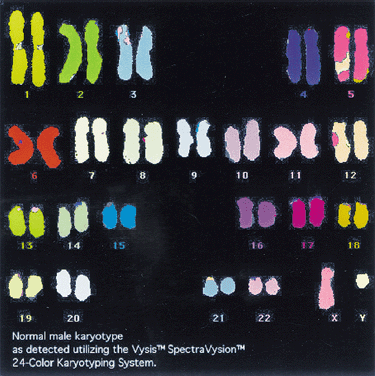
CONCLUSIONS
The role of cytogenetics in obstetrics and gynecology increased dramatically during the past three decades as a consequence of a series of remarkable technologic advancements. Analysis of the structure and behavior of human chromosomes now has direct effect on the diagnosis, management, treatment, prognosis, and prevention of genetic disorders. Cytogenetic studies showed the significant contribution of chromosome aberrations to infertility, pregnancy loss at all stages of gestation, and developmental disabilities and congenital malformations in the newborn. The study of human chromosomes also contributed to the understanding of the relation of genetics to the origin of cancers, the physical mapping of genes, and the prevention of genetic diseases by prenatal testing.
In the first decade (1959 to 1968), cytogenetic analysis based on nonbanded chromosomes led to the following:
The chromosome cause of previously described syndromes was defined (e.g. Down, Klinefelter, and Turner syndromes).
New syndromes were identified as being caused by chromosome aberrations (e.g. trisomy 18).
More than 150 different chromosome aberrations were described by 1964.
The possibility of prenatal diagnosis was raised, and tissue culturing of amniotic fluid cells was begun.
The role of chromosome aberrations in the cause of spontaneous abortions was shown.
The mutagenic potential of radiation, chemicals, and viruses was evaluated by means of chromosome analysis.
The relation of chromosome aberrations to the origin of cancer was suggested by the consistent presence of the Philadelphia (Ph) chromosome in patients with chronic myelogenous leukemia.
In the second decade (1969 to 1978), the introduction of chromosome banding techniques expanded the utility of cytogenetic analysis. Previously established chromosome syndromes were confirmed; many entirely new chromosome syndromes was identified that could not be detected with nonbanded chromosomes; improved chromosome analysis led to the finding that over 125 structural aberrations consistently were associated with specific malignancies; and in conjunction with chromosome analysis, somatic hybridization initiated the mapping of human genes on specific chromosomes.
In the third decade (1979 to 1989), the development of high-resolution banding techniques improved the level of resolution and therefore the amount of clinical information provided by karyotyping. Microdeletions contributing to human pathology were discovered, particularly in association with a broad spectrum of malignancies.
In the fourth decade (1990 to 2000), the introduction of molecular in situ hybridization further expanded the resolution of the human chromosome complement and, applied to interphase cells, made possible more rapid and accurate diagnoses of chromosome disorders in the 1990s. Molecular cytogenetic techniques have been successfully applied to the analysis of fetal cells isolated from maternal blood112 and to the diagnosis of chromosome aberrations in preimplantation embryos.113
In the next decade, it is anticipated that conventional chromosome analysis requiring culturing, harvesting, staining, and microscopic examination will be completely replaced by molecular techniques involving DNA “chips.” A complete and accurate chromosome analysis of single cells will be possible within minutes of sampling based on recombinant DNA technology.
REFERENCES
Conner JM, Ferguson-Smith MA: Essential Medical Genetics. Oxford: Blackwell Scientific Publications, 1987 |
|
Plachot M, Veiga A, Montagut J et al: Are clinical and biological IVF parameters correlated with chromosomal disorders in early life: A multicentric study. Hum Reprod 3: 627, 1988 |
|
Denver Conference: A proposed standard system of nomenclature of human mitotic chromosomes. Lancet 1:1063, 1960 |
|
Caspersson T, Zech L, Johansson C, Modest EJ: Identification of human chromosomes by DNA-binding fluorescing agents. Chromosoma 30: 215, 1970 |
|
Paris Conference, 1971: Standardization in human cytogenetics. Birth Defects 8(7), 1972 |
|
Simpson JL, Martin AO: Cytogenetic nomenclature. Am J Obstet Gynecol 128: 167, 1977 |
|
Seabright M: A rapid banding technique for human chromosomes. Lancet 2: 971, 1971 |
|
Dutrillaux B, Lejeune J: Sur une novelle technique d'analyse du caryotype human. CR Acad Sci D (Paris) 272: 2638, 1972 |
|
Kanda N: Banding pattern observed in human chromosomes by the modified BSG technique. Hum Genet 31: 283, 1976 |
|
Bloom SE, Goodpasture C: An improved technique for selective silver staining of nucleolar organizer regions in human chromosomes. Hum Genet 34: 199, 1972 |
|
Sanchez O, Yunis JJ: New chromosome techniques and their medical applications. In Yunis JJ (ed): New Chromosomal Syndromes. New York: Academic Press, 1977 |
|
Schwartz S, Palmer CG: High-resolution chromosome analysis: I. Applications and limitations. Am J Med Genet 19: 291, 1984 |
|
Ledbetter DH, Riccardi VM, Airhart SD et al: Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med 304: 325, 1981 |
|
Francke U, Holmes LB, Atkins L, Riccardi V: Aniridia-Wilm's tumor association: Evidence for specific deletion of 11p13. Cytogenet Cell Genet 24: 185, 1979 |
|
Yunis JJ, Ramsay N: Retinoblastoma and subband deletion of chromosome 13. Am J Dis Child 132: 161, 1978 |
|
Kunkel LM: The Wellcome Lecture, 1988. Muscular dystrophy: A time of hope. Proc R Soc Lond B Biol Sci 237: 1, 1989 |
|
Sciorra LJ, Hux C, Day-Salvadore D et al: Trisomy 5 mosaicism detected prenatally with an affected liveborn. Prenat Diagn 12: 477, 1992 |
|
Shepard TH: Catalog of Teratogenic Agents. 7th ed. Oral Contraceptives. Baltimore: Johns Hopkins University Press, 1972 |
|
Fryns JP, Kleczkowska A: Chromosome triploidy. In: Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Uchida IA, Freeman VC: Triploidy and chromosomes. Am J Obstet Gynecol 151: 65, 1985 |
|
Simpson JL, Dische R, Morillo-Cucci G, Connolly GF: Triploidy (69,XXY) in a liveborn infant. Ann Genet 15: 103, 1972 |
|
Wertelecki W, Graham JM, Sergovich FR: The clinical syndrome of triploidy. Obstet Gynecol 47: 69, 1976 |
|
Wake N, Fujino T, Hoshi S et al: The propensity to malignancy of dispermic heterozygous moles. Placenta 8: 319, 1987 |
|
Dodson MG: New concepts and questions in gestational trophoblast disease. J Reprod Med 28: 741, 1983 |
|
Scarbrough PR: Chromosome tetraploidy. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Chaganti RSK: The significance of chromosome change in neoplastic development. In German J (ed): Chromosome Mutation and Neoplasia. New York: Alan R Liss, 1983 |
|
Kaslan MB, Zhan Q, El-Deiry WS et al: A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71: 587, 1992 |
|
Davee MA, Weaver DD: Turner syndrome. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Schnizel A: Catalogue of Unbalanced Chromosome Aberrations in Man. Berlin: Walter de Gruyter, 1984 |
|
Bergsma D (ed): The National Foundation March of Dimes: Birth Defects Compendium. 2nd ed. New York: Alan R Liss, 1982 |
|
Buyse ML: Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Fryns JP: Chromosome 21, trisomy 21. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Borgaonkar DS: Chromosome X, triplo-X. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Borgaonkar DS: Chromosome X, poly-X. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Buehler BA, Huseman CA, Sanger W: Klinefelter syndrome. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Jacobs PA, Brunton M, Melville MM et al: Aggressive behavior, mental subnormality, and the XYY male. Nature 208: 1351, 1965 |
|
Hook EB: Behavioral implications of XYY. Science 179: 139, 1973 |
|
Ratcliffe SG, Tierney I, Nshabo J: The Edinburgh study of growth and development of children with sex chromosome abnormalities. Birth Defects 18: 41, 1982 |
|
Moore CY, Weaver DD: Chromosome XYY. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
LeBeau MM, Rowley JD: Recurring chromosomal abnormalities in leukemia and lymphoma. Cancer Surveys 3: 371, 1985 |
|
Warburton D: De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis. Am J Hum Genet 49: 995, 1991 |
|
Kelly TE: Clinical Genetics & Genetic Counseling. 2nd ed. Chicago: Year Book Medical Publishers, 1986 |
|
Steinberg C, Zackai EH, Eunpul DL et al: Recurrence rate for de novo 21q21q translocation Down syndrome: A study of 112 families. Am J Med Genet 17: 523, 1984 |
|
Schmickel RD: Contiguous gene syndromes: A component of recognizable syndromes. J Pediatr 109: 231, 1986 |
|
Pfeiffer RA: Langer-Giedion syndrome and additional malformations with interstitial deletion of the long arm of chromosome 8: Karyotype: 46,XY, del(8)(q13–22). Clin Genet 18: 142, 1980 |
|
Riccardi VM, Sujansky E, Smith AC et al: Chromosomal imbalance in the aniridia-Wilms tumor association: 11p interstitial deletion. Pediatrics 61: 604, 1978 |
|
Knudson AG, Meadows AT, Nichols WW, Hill RR: Chromosomal deletion and retinoblastoma. N Engl J Med 295: 1120, 1976 |
|
van Tuinen P, Dobyns WB, Rich DC: Molecular detection of microscopic and submicroscopic deletions associated with Miller-Dieker syndrome. Am J Hum Genet 43: 587, 1988 |
|
Driscoll DA, Budarf HL, Emanuel BS: A genetic etiology for DiGeorge syndrome: Consistent deletion and microdeletions of 22q11. Am J Hum Genet 50: 924–33, 1992 |
|
Monaco AP, Kunkel LM: Cloning of the Duchenne/Becker muscular dystrophy locus. Adv Hum Genet 17: 61, 1988 |
|
Riggins GJ, Lokey LK, Chastain JL et al: Human genes containing polymorphic trinucleotide repeats. Nat Genet 2: 186, 1992 |
|
Kremer EJ, Pritchard M, Lynch M et al: DNA instability at the fragile X maps to a trinucleotide repeat sequence p(CCG)n. Science 252: 1711, 1991 |
|
Connor JM, Ferguson-Smith MA: Essential Medical Genetics: Chromosomal Disorders. Oxford: Blackwell Scientific Publications, 1987 |
|
Lyon MF: The William Allan Memorial Award Address: X-chromosome inactivation and the location and expression of X-linked genes. Am J Hum Genet 42: 008, 1988 |
|
Latt SA: Microfluorometric analysis of DNA replication in human X chromosomes. Exp Cell Res 86: 412, 1974 |
|
Gartler SM, Dyer KA, Goldman MA: Mammalian X chromosome inactivation. In Friedmann T (ed): Molecular Genetic Medicine. Academic Press, New York. 2: 121, 1992 |
|
Brown CJ, Ballabio A, Rupert JL et al: A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349: 38–44, 1991 |
|
Richler C, Soreq H, Wahrman J: X inactivation in mammalian testis is correlated with inactive X-specific transcription. Nat Genet 2: 192, 1992 |
|
Salido EC, Yen PH, Mohandas TK, Shapiro LJ: Expression of the X-inactivation-associated gene XIST during spermatogenesis. Nat Genet 2: 196, 1992 |
|
McCarrey JR, Dilworth DD: Expression of Xist in mouse germ cells correlates with X-chromosome inactivation. Nat Genet 2: 200, 1992 |
|
Goldman MA: The silence of the X. Nat Genet 2: 169, 1992 |
|
Foote S, Vollrath D, Hilton A, Page DC: The human Y chromosome: Overlapping DNA clones spanning the euchromatic region. Science 258: 60, 1992 |
|
Vollrath D, Foote S, Hilton A: The human Y chromosome: A 43-interval map based on naturally occurring deletions. Science 258: 52, 1992 |
|
Simpson JL: Chromosome mosaicism, 45,X/46,XY type. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1990 |
|
Chang HJ, Clark RD, Bachman H: The phenotype of 45,X/46,XY mosaicism: An analysis of 92 prenatally diagnosed cases. Am J Hum Genet 46: 156, 1990 |
|
Simpson JL: Gonadal dysgenesis, XY type. In Buyse ML (ed): Birth Defects Encyclopedia. Cambridge: Blackwell Scientific Publications, 1992 |
|
Page DC, Mosher R, Simpson EN et al: The sex-determining region of the human Y chromosome encodes a finger protein. Cell 51: 1091, 1987 |
|
Verkerk AJMH, Pieretti M, Sutcliffe JS et al: Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905, 1991 |
|
Richards RI, Sutherland GR: Dynamic mutations: A new class of mutations causing human disease. Cell 70: 709, 1992 |
|
Bloom AD: Induced chromosomal aberrations in man. In Harris H, Hirschhorn K (eds): Advances in Human Genetics. New York: Plenum Press, 1972 |
|
Hook EB, Chambers GM: Estimated rates of Down syndrome in live births by one year maternal age intervals for mothers aged 20–49 in a New York state study: Implications of the risk figures of genetic counseling and cost-benefit analysis of prenatal diagnosis programs. Birth Defects 13: 123, 1977 |
|
Dellarco VL, Voytek PE, Holbender A (eds): Aneuploidy: Etiology and Mechanism. Basic Life Sciences, vol 36. New York: Plenum Press, 1985 |
|
Bender M, Awa A, Brooils A et al: Current status of cytogenetics procedures to detect and quantify previous exposures to radiation. Mutat Res 196: 103, 1988 |
|
Martin RH, Rademaker A, Hildebrand K et al: Variations in the frequency and type of sperm chromosomal abnormalities among normal men. Hum Genet 77: 108, 1987 |
|
Plachot M, de Grouchy J, Junca AM et al: Chromosome analysis of human oocytes and embryos: Does delayed fertilization increase chromosome imbalance? Hum Reprod 3: 125, 1988 |
|
Plachot M, De Grouchy J, Cohen J, Salat-Baroux J: Chromosome abnormalities of the fertilized human egg. Reprod Nutr Dev Suppl 1: 83s, 1990 |
|
Carr DH, Gedeon M: Population cytogenetics of human abortuses. In Hook EB, Porter IH (ed): Population Cytogenetics: Studies in Human Reproduction. New York: Academy Press, 1977 |
|
Ledbetter DH, Martin AO, Verlinsky Y, et al: Cytogenetic results of chorionic villus sampling: High success rate and diagnostic accuracy in the United States collaborative study. Am J Obstet Gynecol 162: 495, 1990 |
|
Alberman ED, Creasy MR: Frequency of chromosomal abnormalities in miscarriages and perinatal deaths. J Med Genet 14: 313, 1977 |
|
Simpson JL: Repeated suboptimal pregnancy outcomes. Birth Defects 17: 1, 1981 |
|
Bourrouillou G, Dastugue N, Colombies P: Chromosome studies in 952 Infertile males with a sperm count below 10 million/ml. Hum Genet 71: 366, 1985 |
|
Kalousek DK, Dill FJ: Chromosomal mosaicism confined to the placenta in human conceptions. Science 211: 665, 1983 |
|
Wilkins-Haug L, Greene M, Roberts DJ, Morton C: Frequency of confined placental mosaicism in pregnancies with intrauterine growth retardation. Am J Obstet Gynecol 166: 350A, 1992 |
|
Kalousek DK, Barrett IJ, Gartner AB: Spontaneous abortion and confined chromosomal mosaicism. Hum Genet 88: 642, 1992 |
|
Kalousek DK, Barrett IJ, McGillivary BC: Placental mosaicism and intrauterine survival of trisomies 13 and 18. Am J Hum Genet 44: 338, 1989 |
|
Hall J: Genomic imprinting: Review and relevance to human diseases. Am J Hum Genet 46: 857, 1990 |
|
Spence JE, Perciaccante RG, Greig GM et al: Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet 42: 217, 1988 |
|
Little M, Van Heyningen V, Hastie N: Dads and disomy and disease. Nature 351: 609, 1991 |
|
Engel E, DeLozier-Blanchet CD: Uniparental disomy, isodisomy, and imprinting: Probable effects in man and strategies for their detection. Am J Med Genet 40: 432, 1991 |
|
Reik WI: Genomic imprinting and genetic disorders in man. Trends Genet 5: 331, 1989 |
|
Haig D, Graham C: Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell 64: 1045, 1991 |
|
Iles S, McBurney W, Bramwell SR et al: Development of parthenogenetic and fertilized mouse embryos in the uterus and in extra-uterine sites. J Embryol Exp Morphol 34: 387, 1975 |
|
Shulman LP, Emerson DS, Felker RE et al: High frequency of cytogenetic abnormalities in fetuses with cystic hygroma diagnosed in the first trimester. Obstet Gynecol 80: 80, 1992 |
|
van Zalen-Sprock RM, van Vugt JMG, van Geijn HP: First-trimester diagnosis of cystic hygroma-course and outcome. Am J Obstet Gynecol 167: 94, 1991 |
|
Jauniaux E, van Maldergen L, Demunter C et al: Nonimmune hydrops fetalis associated with genetic abnormalities. Obstet Gynecol 75: 568, 1990 |
|
Brady K, Polzin WJ, Kopelman JN, Read JA: Risk of chromosomal abnormalities in patients with idiopathic polyhydramnios. Obstet Gynecol 79: 234, 1992 |
|
Chan L, Hixson JL, Laifer SA et al: A sonographic and karyotypic study of second-trimester fetal chorioid plexus cysts. Obstet Gynecol 73: 703, 1989 |
|
Banacerraf BR, Mandell J, Estroff JA et al: Fetal pyelectasis: A possible association with Down syndrome. Obstet Gynecol 76: 58, 1990 |
|
Speleman F, de Potter C, Dal Cen P et al: I(12p) in a malignant ovarian tumor. Cancer Genet Cytogenet 45: 49, 1990 |
|
Hoffner L, Shen-Schwarz S, Deka R et al: Genetics and biology of human ovarian teratomas. III. Cytogenetics and origins of malignant ovarian germ cell tumors. Cancer Genet Cytogenet 62: 58, 1992 |
|
DG Harden, HP Klinger (eds): An International System of Human Cytogenetic Nomenclature (1985): Report of the Standing Committee on Human Cytogenetic Nomenclature. New York: Karger, 1985 |
|
Medical Research Council European trial of chorion villus sampling. MRC working party on the evaluation of chorion villus sampling. Lancet 337:1491, 1991 |
|
Tkachuk DC, Pinkel D, Kuo W-L et al: Clinical applications of fluorescence in situ hybridization. Genet Anal Tech Appl 8: 67, 1991 |
|
Sawyer JR, Johnson MP, Miller OJ: Traditional and molecular cytogenetics. J Reprod Med 37: 485, 1992 |
|
Pinkel D, Straume T, Gray JW: Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA 83: 2934, 1986 |
|
Lichter P, Boyle AL, Cremer T, Ward DC: Analysis of genes and chromosomes by nonisotopic in situ hybridization. Genet Anal Tech Appl 8: 24, 1991 |
|
Klinger K, Landes G, Shook D et al: Rapid detection of chromosome aneuploidies in uncultured amniocytes by using fluorescence in situ hybridization (FISH). Am J Hum Genet 51: 55, 1992 |
|
Lawrence JB, Villnave CA, Singer RH: Sensitive, high resolution chromatin and chromosome mapping in situ: Presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell 52: 51, 1988 |
|
Garson JA, van den Berge JA, Kemshead JT: Novel nonisotopic in situ hybridization technique detects small (1 kb) unique sequences in routinely G-banded human chromosomes: Fine mapping of N-myc and B-NGF genes. Nucleic Acids Res 15: 4761, 1987 |
|
Kallioniemi OP, Kallioniemi A, Sudar D et al: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258: 818, 1992 |
|
Schrock E, du Manoir S, Veldman T et al: Multicolor Spectral Karyotyping of Human Chromosomes. Science 273: 494, 1996 |
|
Elias S, Price J, Dochler M et al: First trimester prenatal diagnosis of trisomy 21 in fetal cells from maternal blood. Lancet 340: 1033, 1992 |
|
Grifo J, Boyle A, Fischer E et al: Pre-embryo biopsy and analysis of blastomeres by in situ hybridization. Am J Obstet Gynecol 163: 2013, 1990 |