Klinefelter Syndrome
Authors
INTRODUCTION
The authors thank Joe Leigh Simpson, MD, for his helpful discussion.
In 1942, Klinefelter and colleagues1 delineated a syndrome, found in nine postpubertal males, consisting of gynecomastia; normal external genitalia; lack of pubertal virilization; small, firm testes with tubular hyalinization, but with a normal number of Leydig cells; azoospermia; elevated gonadotropin levels; and decreased 17-ketosteroid levels. Subsequently, several groups reported that patients with the Klinefelter phenotype were often X-chromatin positive, and the presence of an extra X chromosome was hypothesized.2,3,4 Jacobs and Strong5 in 1959 showed that an X-chromatin–positive male had the complement 47,XXY.
This chapter reviews various aspects of Klinefelter syndrome, including epidemiology, cytogenetics, molecular genetics, clinical manifestations, and management. The discussion generally follows that of Simpson.6 In this chapter, the minimal diagnostic criterion for this syndrome is the presence of at least one Y chromosome and two X chromosomes (Fig. 1).
|
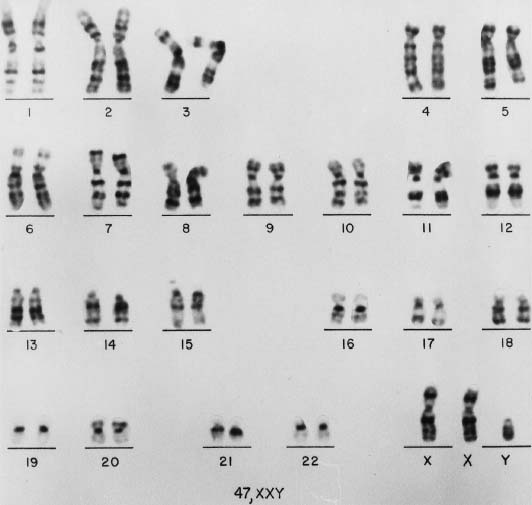
CELLULAR ORIGIN OF KLINEFELTER’S SYNDROME
The most frequent chromosomal complement associated with Klinefelter syndrome is 47,XXY, which may result from a nondisjunction during (1) meiosis I or meiosis II of oogenesis, or (2) meiosis I of spermatogenesis. By using X-linked traits such as Xg blood type or color blindness as markers, it is sometimes possible to determine whether the nondisjunction is of maternal or paternal origin.7 Using polymorphic DNA markers, several investigators have found that the extra X chromosome is of paternal origin in 50% to 60% of cases and of maternal origin in the remaining 40% to 50% of cases.8,9,10 Among the maternal cases, nondisjunction is due to an error in meiosis I division in 70% of cases, an error in meiosis II division in 20% of cases, and a postzygotic mitotic error in 10%.9,10,11 Increased maternal age has been demonstrated in approximately 75% of cases due to maternal meiosis I nondisjunction.11 In the paternally derived 47,XXY cases, the X-Y nondisjunction must always occur in the first meiotic division. Hassold and colleagues,12 in a study of 41 paternally derived 47,XXY cases, found evidence of reduced recombination within the pseudoautosomal region to be associated with X-Y nondisjunction.
Mosaicism of the karyotype 46,XY/47,XXY, which accounts for approximately 15% of Klinefelter syndrome cases, probably arises by nondisjunction or anaphase lag in the zygote or embryo. It is usually assumed that such an error involved a 47,XXY embryo.7
Turning to less common forms of Klinefelter syndrome, the etiology of 49,XXXXY (more than 100 cases documented) is best explained by successive nondisjunction at either meiosis I or II of oogenesis.7 Postzygotic errors, however, may be equally important.
The etiology of the 48,XXYY chromosomal complement (more than 60 cases recorded) may best be explained by either successive nondisjunction at both the first and second meiotic division during spermatogenesis and syngamy of the resulting 25,XYY spermatozoon with a 23,X ovum, or nondisjunction at the second meiotic division in both parents so that syngamy occurs between a 24,XX ovum and a 24,YY sperm.
MOLECULAR MECHANISM
Studies on phenotype-genotype correlations in Klinefelter syndrome patients have been limited. Although there appear to be no obvious phenotypic differences between cases in which the extra X chromosome is of paternal or maternal origin, a minority of Klinefelter syndrome patients are clinically and intellectually much more adversely affected than the average 47,XXY patients. Studies involving structurally abnormal X chromosomes suggest that the Klinefelter’s phenotype is associated with genes mapping to the long arm of the X chromosome.13 Zang14 hypothesized that a nondisjunction error in meiosis II or during postzygotic division leading to two copies of the same X chromosome could induce a negative dosage effect of some harmful recessive genes on the X chromosome. Jacobs and colleagues8 further postulated an imprinting effect on the phenotype of the patients, depending on the parental origin of the extra chromosome. In a study of 80 females with Turner’s syndrome (45,X), Skuse and colleagues15 demonstrated that the presence of an imprinted locus on the long arm of the X chromosome plays a role in cognitive and social development. Interestingly, as discussed below, Klinefelter syndrome patients display behavior problems and impaired language skills, suggesting that imprinted genes localized to the X chromosome are likely to be involved in the observed phenotypic variability. However, no studies have yet demonstrated a distinct difference in phenotype between the paternally and maternally derived cases.
EPIDEMIOLOGY
X-chromatin surveys of unselected newborn boys have revealed an incidence of X-chromatin–positive males ranging from 1.1:1000 to 1.9:1000.16,17,18 Jacobs19 tabulated data from various centers and found that 37 (0.09%) of 37,557 male newborns had 47,XXY complements. Because some males with X chromatin are 46,XX or 46,XY/47,XXY, X-chromatin surveys and chromosomal surveys are not necessarily comparable.
Increased maternal age is associated with an additional X chromosome in both males and females.20 In a series of 45 cases of 47,XXY patients, Ferguson-Smith and colleagues21 found an average maternal age of 32.5 years, compared with 28.7 years for controls and 36.1 years for mothers of children with Down’s syndrome. Hamerton22 reviewed the parental age distribution for two separate series of 47,XXY patients and found a far higher proportion of both mothers and fathers over age 35 than would be expected in the general population of either Sweden or Great Britain. Patil and colleagues23 calculated that the highest risk of having 47,XXY offspring is to mothers over age 40; Carothers and Filippi24 reported that at age 40, the risk is two to three times that of a mother at age 30.
Klinefelter syndrome accounts for 10% to 20% of males attending infertility clinics.25 Mental retardation is a feature in a small percentage of patients with Klinefelter syndrome, as indicated by the increased prevalence of males with X chromatin among residents of institutions for the mentally retarded, compared to the general population.22 The prevalence is also greater among institutionalized patients with psychotic or neurotic disorders and/or a criminal history.26,27,8,29
PATHOLOGY AND CLINICAL MANIFESTATIONS
47,XXY
GENITAL AND GONADAL PATHOLOGY.
External and Internal Genitalia.
In Klinefelter syndrome, the external genitalia are usually well differentiated (Fig. 2), but development of the penis and scrotum may be delayed. Problems of penile development in the first 12 to 14 weeks’ gestation can give rise to incomplete morphogenesis associated with anomalies such as hypospadias, chordee, and micropenis. Problems after 14 weeks’ gestation usually result in a well-formed but unusually small penis.30 The penis is normal in size in 80% to 90% of 47,XXY patients31,32; nevertheless, 47,XXY Klinefelter syndrome is the most commonly known testicular disorder associated with micropenis. The cause is probably a moderate deficiency in testosterone production,33 and the penis is only occasionally incomplete in morphogenesis. The prostate is smaller than usual, also presumably owing to decreased testosterone levels.34 Cryptorchidism is infrequent.
|
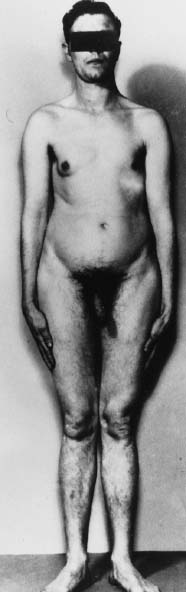
Testes.
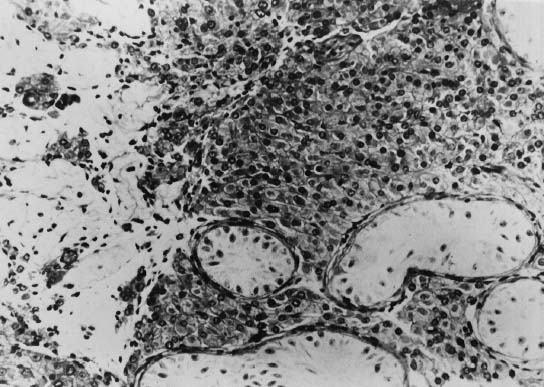
Although the prepubertal testes of 47,XXY patients are usually described as normal in size, Laron and Hochman35 reported that even before puberty the testes are smaller than those of normal boys. Prepubertal 47,XXY patients display neither seminiferous tubule atrophy nor apparent Leydig cell hyperplasia, but primary spermatogonia are reduced in number.36,37 The testes of 47,XXY adults are rarely more than 2 cm in greatest diameter, compared with 3.5 cm in normal males. The testes are usually firmer than normal but are sometimes softer.32 The differences in consistency probably reflect differences in the extent of seminiferous tubule hyalinization. The typical histologic pattern of the adult testes of 47,XXY patients consists of hyalinization and fibrosis of the seminiferous tubules and a relative increase in the number of Leydig cells, with variable degrees of clumping (Fig. 3). Interstitial areas also contain increased numbers of fibroblasts and fat cells.38 Leydig cells are abnormal both histologically and functionally: histologically, they lack crystalloids of Reinke; functionally, their steroid production is decreased.39 Abnormal Leydig cell function is evidenced by decreased plasma testosterone, subnormal response to administration of human chorionic gonadotropin, and increased gonadotropin levels.40 Azoospermia has been reported in at least 90% of these patients, and except for a few instances, paternity has not been established.41 It is quite likely that most if not all cases of paternity in these patients represent instances of undetected mosaicism.
|
SECONDARY SEXUAL DEVELOPMENT.
Klinefelter syndrome becomes more conspicuous in adolescence. There is a delay in the onset of puberty in about half of 47,XXY patients, and reduced penile size in about 20%.42 Pubic hair is usually feminine in distribution; facial and body hair are scanty, and few patients shave daily. Adolescent acne is rare, and temporal hair recession usually does not occur. Obesity, gynecoid fat distribution, and poor muscular development are also frequent. Sexual activity is often reduced,43 although libido and potency may be improved with testosterone therapy.44
Increased parenchymal breast tissue, as determined by palpation, is found in 50% to 75% of 47,XXY patients,39,45 but only approximately 20% have overt gynecomastia.32 Breast enlargement in 47,XXY patients is characterized by increased collagenous material in interglandular spaces; the ductal epithelium is only slightly hyperplastic.6
ENDOCRINE STUDIES.
Prepubertal 47,XXY boys show no significant abnormalities in gonadotropin or testosterone serum levels.44 In adults with 47,XXY complements, levels of both follicle-stimulating hormone (FSH) and luteinizing hormone (LH) are usually elevated,40,45 and testosterone concentrations are usually below normal or in the lower part of the normal range (5 to 86 mg/100 mL).46 The gonads are the principal source of testosterone in patients with Klinefelter syndrome, as in normal males. The total plasma testosterone level, however, is affected by peripheral metabolism, alterations in binding proteins, and diurnal changes and may not be a sensitive indicator of testicular androgen synthesis. These factors contribute to variable reports regarding the plasma testosterone levels in Klinefelter syndrome patients.
As noted earlier, both plasma LH and FSH values are elevated in Klinefelter syndrome patients. These elevations result from the deficient testicular function and the consequent absence of feedback inhibition by sex steroids and other testicular products (e.g., inhibin), probably acting at both the pituitary and hypothalamic levels.47,48 Smals and colleagues49 found elevated gonadotropins that responded to a bolus of LH-releasing hormone (LHRH). In addition, an 8-hour infusion of LHRH elicited an increase in testosterone as well, indicating that despite the resting hypogonadism, the Leydig cells in Klinefelter syndrome have a functional reserve.
Abnormal glucose tolerance test (GTT) results and diabetes mellitus are more frequent among Klinefelter syndrome patients and their parents than in the general population.50,51 Of 157 cases of Klinefelter syndrome reviewed by Nielsen,51 28 (29%) had a diabetic GTT result and 13 (18%) had overt diabetes mellitus, compared to a 6.2% incidence of an abnormal GTT result in a random population less than 50 years of age and 16.1% in those older than 50. Diabetes mellitus is usually mild in Klinefelter syndrome patients.51 Engelberth and colleagues52 reported high levels of autoantibodies against insulin and against pancreatic tissue in some patients with Klinefelter syndrome. Nielsen,51 however, found no such antibodies in his Klinefelter syndrome patients with diabetes mellitus.
Although clinical hypothyroidism is rare, decreased 131I uptake and poor responsiveness to thyroid-stimulating hormone have been reported.53,54 Thyroid-binding globulin levels are usually normal,55 and antithyroid antibodies are usually absent.55
SOMATIC ANOMALIES.
The height of 47,XXY boys under age 3 falls within normal distributions. After age 3, however, the height distribution is skewed, with significantly fewer boys than expected having height percentiles below 25.56 Adult 47,XXY patients are frequently taller than average and often have abnormal skeletal proportions, with relatively long legs and a decreased upper:lower segment ratio.56,57 Klinefelter syndrome patients differ from other eunuchoid patients in that their arm span is usually only 2 to 3 cm greater than their height, if at all greater; in most other types of eunuchoidism, the arm span is usually at least 4 cm greater than the height. The cellular cause of the abnormal proportions is unknown. It is possible that the epiphyses respond abnormally to androgens,39 or an early acceleration of growth rather than a later epiphyseal closure may cause excessive length in the lower limbs. The epiphyses, in fact, close earlier in Klinefelter syndrome boys than in girls.58 The bone age is usually normal.
Robinson and colleagues59 summarized the findings of 63 neonates with 47,XXY complement prospectively ascertained through various chromosome surveys of unselected newborns. In 18% of these boys, one or more major congenital abnormalities were found:
Cleft palate: four cases
Inguinal hernia: four cases
Undescended testes: four cases
Unilateral kidney agenesis and ureter deformities: one case
Microcephaly: one case
Corneal opacity: one case
Aortic stenosis: one case
Nerve deafness: one case
Hypospadias: one case
Funnel chest: one case
Small forehead: one case.
Minor anomalies (e.g., clinodactyly, squint, external rotation of legs, third fontanel, small penis, antimongoloid slants, recurvated knees) were found in 26% of the boys. Of 224 siblings, only 2 (1%) had major anomalies; minor abnormalities were found in 15 (7%).
Simpson and colleagues60 have tabulated from the literature a comparison of some clinical features of 47,XXY, 48,XXXY, and 49,XXXXY patients (Table 1). These tabulations suggest that somatic anomalies are more frequent in 49,XXXXY than in 48,XXXY and even less frequent in 47,XXY. The spectrum of anomalies in 48,XXXY appears to be the same as the spectrum in 49,XXXXY, although anomalies occur less often. The most frequent skeletal anomalies in 47,XXY (e.g., scoliosis, kyphosis, pectus excavatum, clinodactyly V) are the same that are frequently present in 48,XXXY and 49,XXXXY patients. The frequency of congenital heart defects may be higher.61,62 No single anomaly, however, is consistently present. Autoimmune diseases, such as systemic lupus erythematosus,63,64,65 ankylosing spondylitis,66 and Sjögren’s syndrome, characterized by keratoconjunctivitis, dry mouth, and arthritis,65 occur with increased frequency in Klinefelter syndrome patients. It has been hypothesized that testosterone protects males from autoimmune phenomena; therefore, hypogonadal males are prone to defects in T-cell activity that lead to autoimmune disorders.65 Chronic leg ulcerations have also been associated with Klinefelter syndrome, with a reported prevalence as high as 13%.67,68,69 Platelet hyperaggregability, rather than venous disease, appears to be the causative factor.69 In addition, Klinefelter syndrome patients are said to be prone to varicose veins and chronic pulmonary diseases (e.g., emphysema, bronchitis, asthma).32,70,71
TABLE 1. Comparison of Some Clinical Features of Patients With 47,XXY, 48,XXXY, and 49,XXXXY Klinefelter’s Syndrome
47,XXY† | 48,XXXY† | 49,XXXXY§ | ||||
Anomaly* | Cases | N | Cases | N | Cases | N |
Mental retardation | 6 | 141∥ | 29 | 29 | 28 | 28 |
Small testes | 143 | 143 | 26 | 26 | 28 | 28 |
| Hypoplastic penis | 11 | 44¶ | 11 | 24 | 24 | 28 |
Gynecomastia | 26 | 44¶ | 9 | 24 | 5 | 10# |
Wide-set eyes | 0 | 143 | 2 | 25 | 20 | 23 |
Epicanthal folds | 1 | 143 | 6 | 25 | 16 | 19 |
Upturned nose | 0 | 143 | 0 | 25 | 5 | 23 |
Low-set ears | 0 | 143 | 1 | 25 | 2 | 21 |
Malformed ears | 0 | 143 | 1 | 25 | 11 | 21 |
Strabismus | 0 | 143 | 2 | 25 | 11 | 21 |
Prognathism | 0 | 143 | 2 | 25 | 8 | 16 |
Webbed neck | 0 | 143 | 2 | 25 | 4 | 19 |
Short neck | 0 | 143 | 0 | 25 | 14 | 14 |
Kyphosis | 1 | 143 | 3 | 25 | 9 | 13 |
Scoliosis | 0 | 143 | 2 | 25 | 4 | 9** |
Radioulnar synostosis | 0 | 143 | 3 | 25 | 8 | 19 |
Abnormal ulnar or radius | 0 | 143 | 0 | 25 | 4 | 19 |
Clinodactyly V | 2 | 143 | 7 | 25 | 23 | 26 |
Coxa valga | 0 | 143 | 1 | 25 | 12 | 14 |
Genu valgum | 0 | 143 | 0 | 25 | 6 | 24 |
Pes planus | 0 | 143 | 1 | 25 | 10 | 24 |
Malformed toes | 0 | 143 | 0 | 25 | 5 | 24 |
Pes cavus | 2 | 143 | 0 | 25 | 0 | 24 |
Wide gap, first and second toes | 0 | 143 | 0 | 25 | 2 | 26 |
Talipes equinovarus | 0 | 143 | 0 | 25 | 2 | 26 |
*An anomaly is listed if, in the data surveyed, it was found in any two persons with the same chromosomal complement. A number of minor radiographic anomalies detected in 49,XXXY patients are not listed.
†Tabulated from patients of Court-Brown et al,93 except as noted.
†Tabulated from the study by Simpson et al.60
§Tabulated from Zaleski et al,109 except as noted.
∥Ascertained in surveys of the mentally retarded.
¶Estimate of Froland.34
#Estimate of Ferguson-Smith92
**Estimate based on radiographic studies tabulated by Zaleski et al.109
N = total sample.
(Simpson JL, Morillo-Cucci G, Horwith M et al: Abnormalities of human sex chromosomes: VI. Monozygotic twins with the complement 48,XXXY. Humangenetik 21:301, 1974)
Certain types of malignancies, particularly germ cell tumors, both gonadal72,73 and extragonadal,74,75 appear to occur with increased frequency in Klinefelter syndrome patients. Lee and Stephens74 suggested that dysgenic germ cells might arrest in their migration along the urogenital ridge and result in formation of mediastinal and retroperitoneal germ cell tumors; there have also been rare reports of cerebral germ cell tumors.75,76 In addition, breast carcinoma is 20 times more frequent among Klinefelter syndrome patients than among normal males; 3% to 4% of males with breast cancer are 47,XXY patients44,77,78 however, only 27 cases of breast cancer in these patients have been reported.79,80 Because of the risk of malignancy, as well as for cosmetic purposes, some investigators have advocated mastectomy if gynecomastia is pronounced.32 Lymphoma81 and leukemia82,83 have been reported in 47,XXY patients as well, although no clear association between these malignancies and Klinefelter syndrome has been established.
INTELLIGENCE AND PSYCHOSOCIAL DEVELOPMENT.
Although 47,XXY patients are more likely to be retarded or socially maladjusted than normal (46,XY) males, the exact risk is uncertain. That 47,XXY patients have an increased probability of being retarded can be deduced from observations that 1% of mentally retarded males have 47,XXY.22,84 The prevalence of 47,XXY is higher among patients with an IQ between 50 and 85 than among those with a lower IQ.32,85 Robinson and colleagues,59 reviewing the data of 63 infants with 47,XXY complements prospectively ascertained in several chromosome surveys of neonates, found the distribution of full-scale IQ levels was significantly skewed to the left, with 29% of the boys having IQ levels below 90. The difference between siblings and controls was significant (p < 0.001); however, performance tests correlated well with siblings and controls. Graham and colleagues86 suggested that low IQ scores in patients with Klinefelter syndrome are the result of specific linguistic deficits, rather than a global mental deficiency. 47,XXY boys demonstrated significant impairment in expressive language skills, causing difficulties in word finding, syntactic production, and narrative formulation. Netley87 found a 10- to 20-point reduction in verbal skills persisting through adolescence82 and into adult life.42 Performance IQ scores, however, did not significantly differ from those of controls.87 Early referral for speech therapy may help prevent reading and writing disabilities.88
Persons with Klinefelter syndrome frequently exhibit certain behavioral peculiarities, such as poor self-motivation, passivity, absence of anxiety, and bland facies.89,90,91 They adapt poorly to new situations and may display inappropriately aggressive behavior when confronted with stressful situations. Few, however, are overtly sociopathic. Among 47,XXY patients studied after being prospectively ascertained in newborn chromosome surveys, Robinson and colleagues59 determined that 32% had delayed emotional development, compared to 9% of the siblings (p < 0.002). Maladjustment to structured school situations was found in 20 of 45 cases (44%) among 47,XXY boys, compared to 32 of 136 cases among siblings and controls (24%) (p < 0.025). Psychiatric problems were no more frequent in 47,XXY boys than in siblings and controls.
46,XY/47,XXY
Approximately 15% of Klinefelter syndrome patients have been found to have two or more chromosomally distinct cell populations, about 9% being 46,XY/47,XXY.92 The clinical expression of these mosaic patients is variable, ranging from a nearly normal male phenotype to a moderate form of Klinefelter syndrome. These 46,XY/47,XXY patients are less likely than 47,XXY patients to have gynecomastia, azoospermia, small testes, or decreased facial or pubic hair.31 It is quite likely that the majority of patients with Klinefelter syndrome with active spermatogenesis or putative fertility were in fact 46,XY/47,XXY mosaics.41,93 Systematic studies on the frequency of somatic abnormalities, intelligence, and psychological status in 46,XY/47,XXY patients have not been performed.
Other forms of mosaicism have been reported, but the data are insufficient to determine whether specific phenotypic features are associated.
48,XXXY
More than 40 cases of 48,XXXY complement associated with the Klinefelter syndrome phenotype have been reported.60,93,94,95 All patients have been mentally retarded, but because most 48,XXXY cases were ascertained in surveys of mentally retarded subjects, one cannot conclude that the degree of mental retardation is proportional to the number of X chromosomes. Somatic anomalies occur more often in patients with 48,XXXY than in those with 47,XXY (Fig. 4 see Table 1). About half of the reported patients have some nongonadal developmental anomaly, the most frequent being gynecomastia, short neck, epicanthal folds, radioulnar synostosis, and clinodactyly V.
|
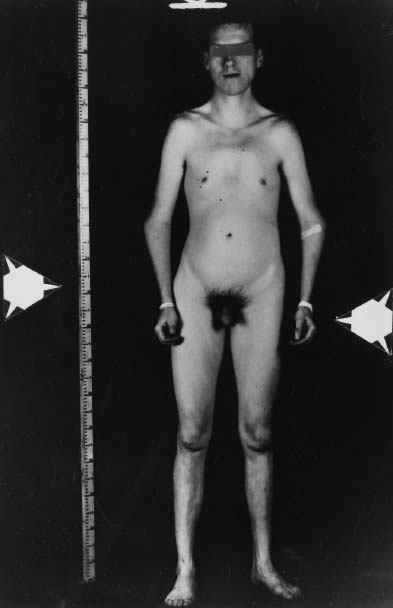
49,XXXXY
More than 100 cases of 49,XXXXY have been reported since Fraccaro and Lindsten96 documented the first example in 1960.97 Most cases were ascertained in surveys of the mentally retarded; none were detected in prospective surveys of newborns. Nearly all patients were severely mentally retarded, with IQ levels usually ranging from 20 to 60.96 In contrast to 47,XXY patients, 49,XXXXY patients often exhibit micropenis and cryptorchidism or incomplete descent of the testes.22 All 49,XXXXY patients have had seminiferous tubule dysgenesis, azoospermia, hypoplastic or absent Leydig cells, and androgen deficiency.98 Additional somatic anomalies are summarized in Table 1.
48,XXYY AND 49,XXXYY
More than 60 patients with 48,XXYY complement have been reported since the first case was published by Muldal and Ockey99 in 1960. Patients with 48,XXYY tend to be taller and more aggressive than those with 47,XXY Klinefelter syndrome.100,101 Otherwise, the phenotype is quite similar to that of 47,XXY. Borgaonkar and colleagues101 reviewed the literature on 53 patients with 48,XXYY complement and found that 62% of the patients over 12 years old had gynecomastia, 18% had micropenis, 12% had cryptorchidism, and 74% had elevated urinary gonadotropins. Testicular histology was similar to that of 47,XXY patients.
Most 48,XXYY patients have been found to be mentally retarded, but because low intelligence was usually the indication for cytogenetic evaluation, ascertainment bias may be a factor. Electroencephalograms are usually abnormal, and epilepsy is not uncommon.101,102 Few of these patients are socially well adjusted, and inappropriate aggression is a common feature. These findings may also reflect ascertainment bias, however, because many cases previously studied were found in institutions for juvenile delinquents.
49,XXXYY has been reported at least twice.103,104 Both patients had mental retardation, delayed bone age, mandibular prognathism, and small testes.
TREATMENT
Medical treatment of patients with Klinefelter syndrome is directed toward the androgen deficiency that is usually present. Smith30 has advocated either intramuscular testosterone cypionate injections (Depo-Testosterone cypionate), 25 mg every 3 weeks for 3 months, or topical application of testosterone cream for the treatment of micropenis in 47,XXY boys. In postpubertal patients with Klinefelter syndrome, androgen replacement therapy produces adequate sexual maturation with increased muscle mass, a more masculine body contour, increased amounts of body hair, enlargement of the penis, hyperpigmentation of the nipples, and advancement of bone age.105 There may also be slight improvement in gynecomastia with androgen therapy.106 Testicular size remains unchanged.105 The long-acting enanthate or the cyclopentylpropionate esters of testosterone are preferred because of their potency and the steady response obtained. The recommended dosage schedule is 200 mg intramuscularly every 1 to 2 weeks for a period of 2 to 3 years, at which time full development is usually attained. Then 100 to 200 mg is administered every 2 to 3 weeks for maintenance. Oral testosterone undecenoate, 40 mg with meals three or four times daily, for long-term substitute treatment has also been recommended.107 Nieschlag108 stated that, because of the shorter half-life, testosterone undecenoate does not suppress a patient’s endogenous testicular androgen production. Alternatively, six 75-mg unconjugated testosterone pellets are placed subcutaneously by means of a pellet injector into the medial aspect of the thighs (three on each side). This procedure must be repeated every 4 to 6 weeks to maintain effective levels of androgen.
Testosterone treatment may also improve behavior and learning disabilities in patients with Klinefelter syndrome. Nielsen and colleagues107 monitored 30 patients after an average of 3.6 years of testosterone therapy and found the majority (77%) had improvement in mood, learning abilities, concentration, energy, and relations with others as compared with their pretreatment status. This conclusion was based on interviews with the patients and their parents and physicians. Although the authors recommended that treatment be started at age 11 or 12, improvement was also noted in the 20- to 30-year-old patients receiving testosterone undecenoate (40 mg with meals); however, many patients in this study preferred Depo-Testosterone injections every 2 to 3 weeks.
Testosterone therapy may also result in improvement in clinical and laboratory features of autoimmune disease in Klinefelter syndrome patients. Bizzarro and colleagues65 described five patients with 47,XXY, three of whom had Sjögren’s syndrome and two of whom had systemic lupus erythematosus. While on oral testosterone undecenoate therapy, serum antinuclear antibody, sedimentation rate, and rheumatoid factor significantly decreased, and the percentage of T-helper cells and T-suppressor/cytotoxic cells significantly increased relative to pretreatment values and values while on placebo. All five patients showed improvement in the clinical features of their autoimmune disorders after initiation of testosterone therapy.
Surgical removal of breast tissue with preservation of the areolae may be necessary, particularly if gynecomastia causes psychological problems. Because of the association between breast cancer and Klinefelter syndrome, all patients with this syndrome should have careful yearly examinations of the breasts.
There is no treatment for the sterility caused by Klinefelter syndrome. Because of their defective development, the testes are unable to respond to either gonadotropic or androgenic stimulation.
GENETIC COUNSELING
The most common indication for prenatal diagnosis is advanced maternal age. Because 47,XXY is associated with both advanced maternal and paternal age, the prenatal diagnosis of a 47,XXY fetus will occasionally be encountered. Other variants associated with Klinefelter syndrome (46,XY/47,XXY; 48,XXXY; 49,XXXXY; 48,XXYY; 49,XXXYY) may also rarely be detected in chorionic villi and amniotic fluid cell cytogenetic studies. In such cases, the couple must be fully informed regarding the current understanding of the potential somatic, social, and psychiatric problems of patients affected with the particular chromosomal abnormality that has been detected. Specifically, few 47,XXY patients are mentally retarded, and relatively few display somatic anomalies or acquired diseases. 46,XY/47,XXY patients are less severely affected than 47,XXY patients. In contrast, all reported 48,XXXY or 49,XXXXY patients have been mentally retarded. The frequency of somatic anomalies is also higher in 48,XXXY than in 47,XXY, and highest of all in 49,XXXXY.109 As in all cases of prenatal diagnosis, the couple must make the ultimate decision about whether or not to continue the pregnancy. Their decision must be supported by the physician and the entire genetic counseling team.
No data are currently available regarding the recurrence risks for 47,XXY or other chromosomal variants of Klinefelter syndrome. Nonetheless, in subsequent pregnancies, we recommend that prenatal diagnosis be offered to couples who have had offspring with Klinefelter syndrome.
REFERENCES
Klinefelter HF Jr, Reifenstein ED Jr, Albright F: Gynecomastia, aspermatogenesis without aleydigism and increased excretion of follicle-stimulating hormone. J Clin Endocrinol 2:615, 1942 |
|
Bradbury JT, Bunge RG, Boccabella RA: Chromatin test in Klinefelter’s syndrome. J Clin Endocrinol Metab 16:689, 1956 |
|
Plunkett ER, Barr ML: Testicular dysgenesis affecting the seminiferous tubules principally, with chromatin-positive nuclei. Lancet 2:853, 1956 |
|
Riis P, Johnsen SG, Mosbeck J: Nuclear sex in Klinefelter syndrome. Lancet 1:962, 1956 |
|
Jacobs PA, Strong JA: A case of human intersexuality having a possible XYY sex-determining mechanism. Nature 183:302, 1959 |
|
Simpson JL: Disorders of Sexual Differentiation. New York, Academic Press, 1976 |
|
Sanger R, Tippett P, Gavin J: Xg groups and sex abnormalities in people of Northern European ancestry. J Med Genet 8:417, 1971 |
|
Jacobs PA, Hassold TJ, Whittington E et al: Klinefelter’s syndrome: An analysis of the origin of the additional sex chromosome using molecular probes. Ann Hum Genet 52:93, 1988 |
|
Harvey J, Jacobs PA, Hassold T, Pettay D: The parental origin of 47,XXY males. Birth Defects 26:289, 1991 |
|
Lorda-Sanchez I, Binkert F, Maechler M et al: Reduced recombination and paternal age effect in Klinefelter syndrome. Hum Genet 89:524, 1992 |
|
MacDonald M, Hassold T, Harvey J et al: The origin of 47,XXY and 47,XXX aneuploidy: Heterogeneous mechanisms and role of aberrant recombination. Hum Mol Genet 3:1365, 1994 |
|
Hassold TJ, Sherman SL, Pettay D et al: XY chromosome nondisjunction in man is associated with diminished recombination in the pseudoautosomal region. Am J Hum Genet 49:253, 1991 |
|
Patil SR, Bartley JA, Hanson JW: Association of the X chromosomal region and Klinefelter syndrome. Clin Genet 19:343, 1981 |
|
Zang KD: Genetics and cytogenetics of Klinefelter syndrome. In Bandmann HJ, Breit R (eds): Klinefelter Syndrome. pp 12, 23 Berlin, Springer, 1984 |
|
Skuse DH, James RS, Bishop DVM et al: Evidence from Turner syndrome of an imprinted X-linked locus affecting cognitive function. Nature 387:705, 1997 |
|
Court-Brown WMS: Sex chromosome aneuploidy in man and its frequency, with special reference to mental subnormality and criminal behavior. Int Rev Exp Pathol 7:31, 1969 |
|
Robinson A, Goad WB, Puck TT et al: Studies on chromosomal nondisjunction in man: III. Am J Hum Genet 21:466, 1969 |
|
Bell AG, Corey PN: A sex chromatin and Y body survey of Toronto newborns. Can J Genet Cytol 16:239, 1974 |
|
Jacobs PA: The incidence and etiology of sex chromosome abnormalities in man. Birth Defects 15:3, 1979 |
|
Court-Brown WM, Law P, Smith PG: Sex chromosome aneuploidy and parental age. Ann Hum Genet 33:1, 1969 |
|
Ferguson-Smith MMA, Mack WS, Ellis PM et al: Parental age and the source of the X chromosomes in XXY Klinefelter syndrome. Lancet 1:46, 1964 |
|
Hamerton JL: Human Cytogenetics. Vol 2:New York, Academic Press, 1971 |
|
Patil SR, Lubs HA, Kimberling WJ et al: Chromosomal abnormalities ascertained in a collaborative survey of 4,342 seven and eight year old children: Frequency, phenotype and epidemiology. In Hook EB, Porter IH (eds): Population Cytogenetics. pp 103, 131 New York, Academic Press, 1977 |
|
Carothers AD, Filippi G: Klinefelter’s syndrome in Sardinia and Scotland. Hum Genet 81:1, 1988 |
|
Summit RL: Cytogenetic disorders. In Jackson LG, Schimke RN (eds): Clinical Genetics: A Source Book for Physicians. pp 35, 84 New York, John Wiley & Sons, 1979 |
|
MacLean N, Court-Brown WM, Jacobs PA et al: A survey of sex chromatin abnormalities in mental hospitals. J Med Genet 5:165, 1968 |
|
Nielsen J: Klinefelter’s syndrome and the XYY syndrome: A genetical, endocrinological and psychiatric-psychological study of thirty-three severely hypogonadal male patients and two patients with karyotype 47,XXY. Acta Psychiatr Scand 45(suppl):209, 1969 |
|
Casey MD, Segall LJ, Street DRK et al: Sex chromosome abnormalities in two state hospitals for patients requiring special security. Nature 209:641, 1966 |
|
Jacobs PA, Price WHH, Court-Brown WM et al: Chromosome studies on men in a maximum security hospital. Ann Hum Genet 31:339, 1968 |
|
Smith DW: Micropenis and its management. Birth Defects 13:147, 1977 |
|
Paulsen CA, Gordon DL, Carpenter RW et al: Klinefelter’s syndrome and its variants: A hormonal and chromosomal study. Recent Prog Horm Res 24:321, 1968 |
|
Becker KL: Clinical and therapeutic experiences with Klinefelter’s syndrome. Fertil Steril 23:568, 1972 |
|
Caldwell P, Smith DW: The XXYY (Klinefelter’s) syndrome in childhood: Detection and treatment. J Pediatr 80:250, 1972 |
|
Froland A: Klinefelter’s syndrome: Clinical, endocrinological, and cytogenetical studies. Dan Med Bulll 16(suppl 6):1, 1969 |
|
Laron Z, Hochman IH: Small testes in prepubertal boys with Klinefelter’s syndrome. J Clin Endocrinol Metabol 32:671, 1971 |
|
Ferguson-Smith MA: The prepubertal testicular lesion in chromatin-positive Klinefelter’s syndrome (primary micro-orchidism) as seen in mentally handicapped children. Lancet 1:219, 1959 |
|
Mikamo K, Aguercif M, Hazeghi P et al: Chromatin-positive Klinefelter’s syndrome. Fertil Steril 19:731, 1968 |
|
Jones HW, Scott WW: Hermaphroditism, Genital Anomalies and Related Disorders. 2nd ed.. Baltimore, Williams & Wilkins, 1971 |
|
Ferguson-Smith MA: Testis and intersexuality. In Hubble D (ed): Paediatric Endocrinology. pp 359, Oxford, Blackwell Scientific, 1969 |
|
Leonard JM, Bremner WJ, Capell PT et al: Male hypogonadism: Klinefelter and Reifenstein syndromes. Birth Defects 11:17, 1976 |
|
Warburg E: A fertile patient with Klinefelter’s syndrome. Acta Endocrinol (Copenh) 43:12, 1963 |
|
Sorenson K: Klinefelter’s syndrome in childhood, adolescence and youth. Carnforth, Parthenon, 1988 |
|
Raboch J: Klinefelter syndrome and sexual development and activity. Arch Sex Behav 8:1, 1979 |
|
Stewart DA, Netley CT, Bailey JD et al: Growth and development of children with X and Y chromosome aneuploidy: A prospective study. Birth Defects 15:15, 1979 |
|
Wang C, Baker WG, Burger HG: Hormone studies in Klinefelter’s syndrome. Clin Endocrinol (Oxf) 4:399, 1975 |
|
Paulsen CA: The testes. In Williams RH (ed): Textbook of Endocrinology. 5th ed.. Philadelphia, WB Saunders, 1974 |
|
Means AR, Fakunding JL, Huckins C et al: Follicle-stimulating hormone, the Sertoli cell, and spermatogenesis. Recent Prog Horm Res 32:477, 1976 |
|
Peterson NT, Midgley AR Jr, Jaffe RB: Regulation of human gonadotropins: III. Luteinizing hormone and follicle stimulating hormone in sera from adult males J Clin Endocrinol Metab 28:1473, 1968 |
|
Smals AGH, Kloppenborg PWC, Lequin RM et al: The effect of gonadotropin releasing hormone on pituitary-gonadal function in Klinefelter’s syndrome. Acta Endocrinol (Copenh) 83:82, 1976 |
|
Nielsen J, Johansen K, Yde H: Frequency of diabetes mellitus in patients with Klinefelter’s syndrome of different chromosome constitutions and the XYY syndrome: Plasma insulin and growth hormone levels after a glucose load. J Clin Endocrinol 29:1062, 1969 |
|
Nielsen J: Diabetes mellitus in patients with aneuploid chromosome aberrations and in their parents. Humangenetik 16:165, 1972 |
|
Engelberth D, Charuat J, Jeykova H: Autoantibodies in chromatin-positive men. Lancet 2:1194, 1965 |
|
Davis TE, Canfield CJ, Herman RH et al: Thyroid function in patients with aspermiogenesis and testicular tubal sclerosis. N Engl J Med 268:178, 1963 |
|
Zuppinger KK, Engel E, Forbes AP et al: Klinefelter’s syndrome: A clinical and cytogenetic study in twenty-four cases. Acta Endocrinol (Copenh) 54(suppl 113):5, 1967 |
|
Carr DH, Barr ML, Plunkett ER et al: An XXXY sex chromosome complex in Klinefelter subjects with duplicated sex chromatin. J Clin Endocrinol 21:491, 1961 |
|
Stewart JSS, Ferguson-Smith MA, Lenox B et al: Klinefelter’s syndrome: Genetic studies. Lancet 2:117, 1958 |
|
Milne JS, Lauder IJ, Price WH: Anthropometry in sex chromosome abnormality. Clin Genet 5:96, 1974 |
|
Stewart JSS: Medullary gonadal dysgenesis (chromatin-positive Klinefelter’s syndrome). Lancet 1:1176, 1959 |
|
Robinson A, Lubs HA, Nielsen J et al: Summary of clinical findings: Profiles of children with 47,XXY, 47,XXX and 47,XYY karyotypes. Birth Defects 15:261, 1979 |
|
Simpson JL, Morillo-Cucci G, Horwith M et al: Abnormalities of human sex chromosomes: VI. Monozygotic twins with the complement 48,XXXY Humangenetik 21:310, 1974 |
|
Gautier M, Nouaille J: Deux cas de syndrome de Klinefelter associe a une tetralogie de Fallot (etude systematique du corpuscule de Barr chez 210 mourrissons attiens decardiopathies congenitales). Arch Fr Pediatr 21:761, 1964 |
|
Rosenthal A: Cardiovascular malformation in Klinefelter’s syndrome: Report of three cases. J Pediatr 80:471, 1972 |
|
Schlegelberger T, Kekow J, Gross WL: Impaired T-cell independent B-cell maturation in systemic lupus erythematosus coculture experiments in monozygotic twins concordant for Klinefelter’s syndrome but discordant for systemic lupus erythematosus. Clin Immunol Immunopathol 40:365, 1986 |
|
Miller MH, Urowitz MB, Gladman DD et al: Systemic lupus erythematosus in males. Medicine 62:327, 1983 |
|
Bizzaro A, Valentini G, Di Martino G et al: Influence of testosterone therapy on clinical and immunological features of autoimmune diseases associated with Klinefelter’s syndrome. J Clin Endocrinol Metab 64:32, 1987 |
|
Armstrong RD, Macfarlane DG, Panavi GS: Ankylosing spondylitis and Klinefelter’s syndrome: Does the X chromosome modify disease expression ? Br J Rheumatol 24:27, 1985 |
|
Norris PG, Rivers JK, Machin S et al: Platelet hyperaggregability in a patient with Klinefelter’s syndrome and leg ulcers. Br J Dermatol 117:107, 1987 |
|
Verp MS, Simpson JL, Martin AO: Hypostatic ulcers in 47,XXY Klinefelter’s syndrome. J Med Genet 20:100, 1983 |
|
Downham TF, Mitek FV: Chronic leg ulcers and Klinefelter’s syndrome. Cutis 38:110, 1986 |
|
Becker KL, Hoffman DL, Albert A et al: Klinefelter’s syndrome: Clinical and laboratory findings in 50 patients. Arch Intern Med 118:314, 1966 |
|
Rohde RA: Klinefelter’s syndrome with pulmonary disease and other disorders. Lancet 2:149, 1964 |
|
Dexeus FH, Logothetisis CJ, Chong C et al: Genetic abnormalities in men with germ cell tumors. J Urol 140:80, 1988 |
|
Carroll PR, Morse MJ, Kodura PP et al: Testicular germ cell tumor in patient with Klinefelter syndrome. Urology 31:72, 1988 |
|
Lee M, Stephens RL: Klinefelter’s syndrome and extragonadal germ cell tumors. Cancer 60:1053, 1987 |
|
Nichols CR, Heerema NA, Palmer C et al: Klinefelter’s syndrome associated with mediastinal germ cell neoplasms. J Clin Oncol 5:1290, 1987 |
|
Arens R, Marcus D, Engelberg S et al: Cerebral germinomas and Klinefelter syndrome. Cancer 61:1228, 1988 |
|
Harnden DG, MacLean N, Langlands AO: Carcinoma of the breast and Klinefelter’s syndrome. J Med Genet 8:460, 1971 |
|
Cuenca CR, Becker KL: Klinefelter’s syndrome and cancer of the breast. Arch Intern Med 121:159, 1968 |
|
Scheike O, Visfeldt J, Petersen B: Male breast cancer: III. Breast carcinoma in association with Klinefelter’s syndrome Acta Pathol Microbiol Scand (A)81:352, 1973 |
|
Evans DB, Crichlow RW: Carcinoma of the male breast and Klinefelter’s syndrome: Is there an association? CA 37:246, 1987 |
|
Becher R: Klinefelter syndrome and malignant lymphoma. Cancer Genet Cytogenet 1:271, 1986 |
|
Ratcliffe SG, Paul N: Prospective studies on sex chromosome aneuploidy. Birth Defects 22, 1986 |
|
Horsman DE, Pantzar JT, Dill FJ et al: Klinefelter’s syndrome and acute leukemia. Cancer Genet Cytogenet 26:375, 1987 |
|
Hambert G: Males with Positive Sex Chromatin. Gotenborg, Sweden, Akademiforlaget, 1966 |
|
Robach J, Sipova I: The mental level in 47 cases of true Klinefelter’s syndrome. Acta Endocrinol (Copenh) 36:404, 1961 |
|
Graham JM, Bashir AS, Stark RE et al: Oral and written language abilities of XXY boys: Implications for anticipatory guidance. Pediatrics 81:6, 1988 |
|
Netley CT: Summary overview of behavioral development in individuals with neonatally identified X and Y aneuploidy. Birth Defects 22:293, 1986 |
|
Editorial review: Klinefelter’s syndrome. Lancet 8598:1316, 1988 |
|
Hoaken PCS, Clarke M, Breslin M: Psychopathology in Klinefelter’s syndrome. Psychosom Med 26:207, 1964 |
|
Federman DD: Abnormal Sexual Development. Philadelphia, WB Saunders, 1967 |
|
Money J, Annecillo C, Van Orman B et al: Cytogenetics, hormones and behavior disability: Comparison of XYY and XXY syndromes. Clin Genet 6:370, 1974 |
|
Ferguson-Smith MA: Klinefelter’s syndrome and mental deficiency. In More KL (ed): Sex Chromatin. p 277, Philadelphia, WB Saunders, 1966 |
|
Court-Brown WM, Harnden DG, Jacobs PA et al: Abnormalities of the Sex Chromosome Complement in Man. Med Res Counc no. 305 London, Her Majesty’s Stationery Office, 1964 |
|
Ferrier PE, Ferrier SA, Pescio G: The XXXY Klinefelter syndrome in childhood. Am J Dis Child 127:104, 1974 |
|
Vormittag V, Weninger W: XXXY Klinefelter’s syndrome. Humangentik 15:327, 1972 |
|
Fraccaro M, Lindsten J: A child with 49 chromosomes. Lancet 2:1303, 1960 |
|
Terheggen HG, Pfeiffer RA, Haug H et al: XXXXY Syndrom. Bericht uber 7 neue Falle and Literaturubersicht Z Kinderheilkd 115:209, 1973 |
|
Cunningham MD, Roigsdale JL: Genital anomalies of an XXXXY male subject. J Urol 107:872, 1972 |
|
Muldal S, Ockey CH: The “double male”: A new chromosome situation in Klinefelter’s syndrome. Lancet 2:492, 1960 |
|
Barlow PW: X-chromosome and human development. Dev Med Child Neurol 15:205, 1973 |
|
Borgaonkar DS, Mules E, Char F: Does the 48,XXYY male have a characteristic phenotype? Clin Genet 1:272, 1970 |
|
Hornstein OP, Rott HD, Schwanitz G et al: Die XXYY-Variante des Klinefelter’s Syndroms. Dtsch Med Wochenscher 99:248, 1974 |
|
Bray P, Hosephine A: An XXXYY-sex chromosome anomaly. JAMA 184:179, 1963 |
|
Lecluse-van der Bilt FA, Hagemeijer A, Smit EM et al: An infant in an XXXYY karyotype. Clin Genet 5:263, 1974 |
|
Myhre SA, Ruvalcaba RHA, Johnson HR et al: The effects of testosterone treatment in Klinefelter’s syndrome. J Pediatr 76:267, 1970 |
|
Fromatin M, Grutier D, Cuisinier JC et al: Resultats de l’androgene-therapie dans le syndrome de Klinefelter de l’adolescent. Ann Endocrinol (Paris) 35:305, 1974 |
|
Nielsen J, Pelsen B, Sorensen K: Followup of 30 Klinefelter males treated with testosterone. Clin Genet 33:4, 1988 |
|
Nieschlag E: Testosterone substitution therapy. In HJ Bandman (ed): Klinefelter’s syndrome. pp 202, 211 New York, Springer-Verlag, 1984 |
|
Zaleski WA, Houston CS, Pozsonyi J et al: The XXXXY chromosome anomaly: Report of three new cases and review of 30 cases from the literature. Can Med Assoc J 94:1143, 1966 |