Production, Clearance, and Measurement of Steroid Hormones
Authors
INTRODUCTION
From the chemical standpoint, hormones can generally be divided into three types: proteins (e.g., FSH and LH), peptides (e.g., GnRH and ACTH), and steroids (e.g., progesterone and estradiol). The basic units of proteins and peptides are amino acids, whereas steroids contain the gonane structure. Proteins are relatively large molecules and are water soluble, whereas steroids are small molecules and are generally soluble in organic solvents. Peptides are somewhere in between these two classes of hormones with respect to molecular weight and solubility.
BASIC STRUCTURE AND CLASSIFICATION OF STEROIDS
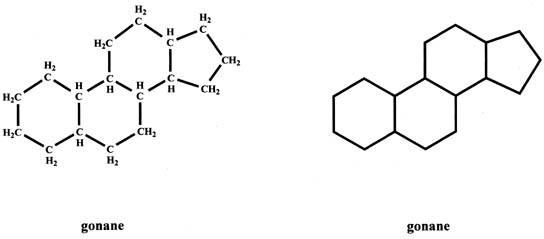
The basic structure of steroids, gonane (cyclopentanoperhydrophenanthrene), has 17 carbons which are arranged as three six-member carbon rings to which a five-member carbon ring is attached (Fig. 1). Each carbon has one or two hydrogens attached. The gonane structure can be represented without showing the carbons and hydrogens, as shown in Figure 1.
|
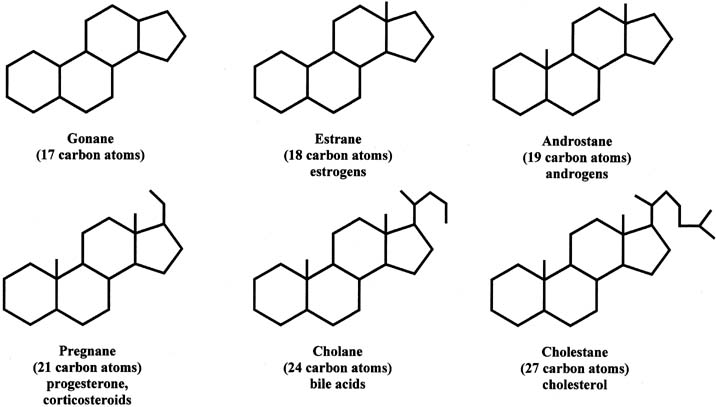
Steroids can be divided into different groups of parent compounds, based on the number of carbons that they contain (Fig. 2). In addition to gonanes, which consist of 17 carbons, estranes consist of 18 carbons (C18 steroids) and include estrogens. Androstanes have 19 carbons (C19 steroids) and include androgens. Pregnanes contain 21 carbons (C21 steroids) and include progesterone and corticosteroids. This chapter focuses primarily on C18, C19, and C21 steroids. Cholanes have 24 carbons and include bile acids, and cholestanes have 27 carbons and include cholesterol as well as cholesterol-like compounds. The compounds in this group are also referred to as sterols.
|
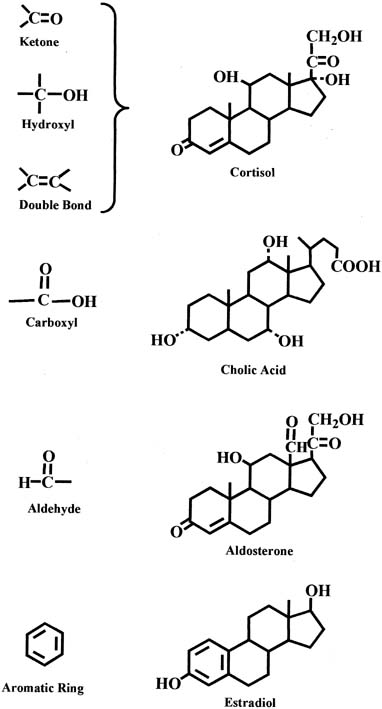
In each group of parent steroids, compounds differ in their characteristics because of the presence of different functional groups on the molecules. Common functional groups include the ketone group, hydroxyl group, and double bond, as shown in the chemical structure of the cortisol molecule in Figure 3. Other functional groups include the carboxyl and aldehyde groups, which are present in the molecules of bile acids and aldosterone, respectively (see Fig. 3). An important characteristic of the C18 steroids is the presence of an aromatic ring that is found in estrogens (e.g., estradiol) (see Fig. 3).
|
FORMATION OF STEROID HORMONES
Sources of steroid hormone formation in the body can be divided into two types (Table 1). One source is the endocrine glands. In women, they include the adrenals, ovaries, and placenta, which is an incomplete endocrine gland. In men, the endocrine glands include the adrenals and testes. A second source of steroid hormones in the body is peripheral tissues. These are nonendocrine tissues such as the liver, intestine, fat, skin, kidneys, and brain.
Table 1. Sources of steroid hormones
| Endocrine Glands |
| In women: adrenals, ovaries, and placenta (incomplete) |
| In men: adrenals and testes |
| Peripheral (Nonendocrine Gland) Tissues |
| Splanchnic, e.g., liver, intestine |
| Extrasplanchnic, e.g., fat, skin (sexual and nonsexual), kidneys, brain, etc. |
The first steroidal precursor for biosynthesis of steroid hormones in the adrenals, ovaries, and testes is cholesterol. In these endocrine glands, cholesterol can be synthesized de novo from acetate by a complex series of reactions. Alternatively, it can be obtained directly from circulating low-density lipoprotein (LDL) cholesterol.
Cholesterol can be converted to a variety of steroid hormones in the endocrine glands through the action of specific enzymes, encoded by different genes. The first and rate-limiting reaction in the formation of steroid hormones is the conversion of cholesterol to pregnenolone, which is stimulated by adrenocorticotropin hormone (ACTH) in the adrenals and by LH in the ovaries and testes. This reaction is complex and occurs in the mitochondria. It is catalyzed by the enzyme C20-22-lyase (also referred to as C20-22-desmolase), which is encoded by the CYP11A gene. A key step in the reaction is the transport of cholesterol from extracellular sources to the inner mitochondrial membrane, and subsequent loading of the precursor into the active site of the enzyme. Intramitochondrial cholesterol movement appears to involve coordinated activation of the steroidogenic acute regulatory (StAR) protein and peripheral-type benzodiazepine receptor.1, 2 Both the delivery of cholesterol to the enzyme and the enzyme level are primarily under the control of tropic hormones (LH or ACTH) using cyclic AMP or calcium as the intracellular messenger. Once pregnenolone is formed, it can then be converted to progesterone, androgens, estrogens, and corticosteroids. For this reason, pregnenolone is sometimes referred to as the “mother” steroid.
Although the adrenals, ovaries, and testes can all synthesize androgens, only the adrenals produce corticosteroids. The ovaries and testes, but not the adrenals, can form estrogens. This does not mean that the adrenals, ovaries, and testes lack the enzymes to synthesize estrogens, or corticosteroids. This is evident in feminizing adrenal tumors, which produce estrone and estradiol in high amounts, and in testicular and ovarian tumors that produce certain corticosteroids. Thus, it appears that the activity of certain steroidogenic enzymes in the adrenals, ovaries, and testes are suppressed by mechanisms that are not yet understood.
The placenta also does not express certain steroidogenic enzymes and, as mentioned previously, is an incomplete endocrine organ. It lacks the enzymes required to form cholesterol, as well as those required to convert progesterone to androgens, and subsequently estrogens.
STEROID HORMONE BIOSYNTHESIS IN THE OVARIES AND TESTES
Figure 4 illustrates the biosynthetic pathways leading to the formation of androgens and estrogens in the ovaries and testes. This occurs by one of two series of biochemical reactions, which are often referred to as the Δ5 pathway and the Δ4 pathway. In the Δ5 pathway, all of the steroids have a double bond between carbons 5 and 6, whereas the double bond is between carbons 4 and 5 in the Δ4 pathway. The relative importance of the two pathways is poorly understood.
|
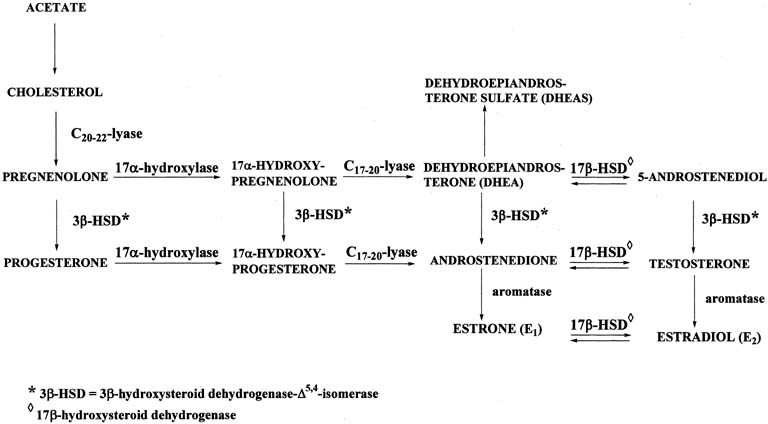
The Δ5 pathway begins by formation of 17-hydroxypregnenolone from pregnenolone via the enzyme, 17α-hydroxylase. Subsequently, 17α-hydroxypregnenolone is converted to dehydroepiandrosterone (DHEA) through the action of C17-20-lyase. Both 17α-hydroxylase and C17-20-lyase reside on a single protein and are encoded by a single gene, namely, CYP17.3, 4 DHEA is then transformed to 5-androstene-3β,17β-diol (androstenediol) by 17β-hydroxysteroid dehydrogenase (also referred to as 17β-hydroxysteroid oxidoreductase [17β-HSD]). As the name oxidoreductase implies, the reaction in which DHEA is converted to androstenediol involves reduction (addition of two hydrogens to the ketone group at carbon 17 of DHEA) or oxidation (removal of two hydrogens from the hydroxyl group at carbon 17 of androstenediol). Thus, the conversion of DHEA to androstenediol is reversible. The Δ5 pathway stops after androstenediol is formed.
The Δ4 pathway begins with the conversion of pregnenolone to progesterone through the action of two enzymes, namely, 3β-hydroxysteroid dehydrogenase and Δ5,4-isomerase. The former enzyme is also referred to as 3β-hydroxysteroid oxidoreductase (3β-HSD). It is encoded by the 3β-HSD gene. There are two functional 3β-HSD genes (type 1 and type 2).5, 6, 7 Type 1 expression occurs primarily in the placenta, mammary gland, and skin, whereas the type 2 isoform is expressed almost exclusively in the adrenals and gonads. The enzymatic reaction involves oxidation, that is, removal of two hydrogens from the hydroxyl group at carbon 3, forming a ketone group. In contrast to the reversible formation of androstenediol from DHEA, this reaction is not reversible to any significant extent. Once the ketone group is formed, the double bond between carbons 5 and 6 is rapidly shifted and becomes located between carbons 4 and 5 through the action of the isomerase enzyme. As shown in Figure 4, in addition to pregnenolone, all of the other compounds in the Δ5 pathway can also be converted to the corresponding Δ4 compounds.
Once progesterone is formed, the reactions in the Δ4 pathway proceed in the same manner as in the Δ5 pathway, with the same enzymes and genes. Thus, progesterone is first converted to 17α-hydroxyprogesterone, which undergoes side-chain cleavage to form androstenedione. This androgen is then transformed to the final product of the Δ4 pathway, which is testosterone, in a reversible reaction catalyzed by the 17β-HSD enzyme.
In humans, there are at least five isoforms of the 17β-HSD enzyme, encoded by the 17β-HSD gene; they are designated types 1 through 5.8 Each type has cell-specific expression, substrate specificity, regulatory mechanisms, and reductase or oxidative catalytic activity. The affinity of the 17β-HSD type 1 isoenzyme is approximately 100-times higher for C18 steroids than for C19 steroids, and its catalytic preference is reduction. It is localized predominantly in the ovary (granulosa cells) and placenta (syncytiotrophoblast). The 17β-HSD type 2 isoenzyme preferentially catalyzes the oxidation of steroids with a hydroxyl group at carbon 17, for example, testosterone, estradiol. The enzyme is distributed among many extraglandular tissues, such as endometrium, placenta, and liver; however, it is primarily expressed in the endometrium. The activity of the type 2 isoenzyme is increased during the luteal phase of the menstrual cycle in a manner that parallels circulating progesterone levels during the cycle. The 17β-HSD type 3 isoenzyme is expressed in the testes and preferentially catalyzes the reduction of androstenedione to testosterone at carbon 17.
Relatively little is yet known about the 17β-HSD type 4 and type 5 isoenzymes. It appears that the type 4 isoenzyme catalyzes the oxidation of C18 steroids, for example, estradiol to estrone, whereas the type 5 isoenzyme catalyzes the reduction of C19 steroids, for example, androstenedione to testosterone.
Deficiency of the 17β-HSD type 3 isoenzyme causes a form of male pseudohermaphroditism referred to as 17β-HSD deficiency, in which there is a defect in the biosynthesis of testosterone from androstenedione in the fetal testes.9 The deficiency is confined to individuals with a 46,XY karyotype. These individuals have testes, wolffian duct-derived male internal genitalia (with the exception of a prostate), female external genitalia, and gynecomastia.
Deficiencies of 17α-hydroxylase and C17-20-lyase in fetal testes have also been reported. As in the disorder of 17β-HSD deficiency, individuals with a defect in one of these enzymes also cannot produce testosterone and have incomplete masculinization.
The two androgens, androstenedione and testosterone, can undergo a series of complex reactions (aromatization) catalyzed by the aromatase enzyme, forming the estrogens, estrone (E1) and estradiol (E2), respectively. This reaction is encoded by the CYP19 gene.10
STEROID HORMONE BIOSYNTHESIS IN THE ADRENALS
Figure 5 shows the biosynthetic pathways of steroid hormone formation, which includes mineralocorticoids, glucocorticoids, and androgens, in the adrenals. In the adrenals, the Δ5 and Δ4 pathways leading to formation of androstenedione and testosterone are the same as those in the ovaries and testes, described in the previous section. Because aromatase activity is not expressed in the adrenals, no estrogens are formed. Instead, the adrenals form corticosteroids. They are formed by the mineralocorticoid and glucocorticoid pathways. The adrenals also have high sulfuryl transfer activity, which catalyzes the formation of DHEAS from DHEA (see Fig. 5).
|
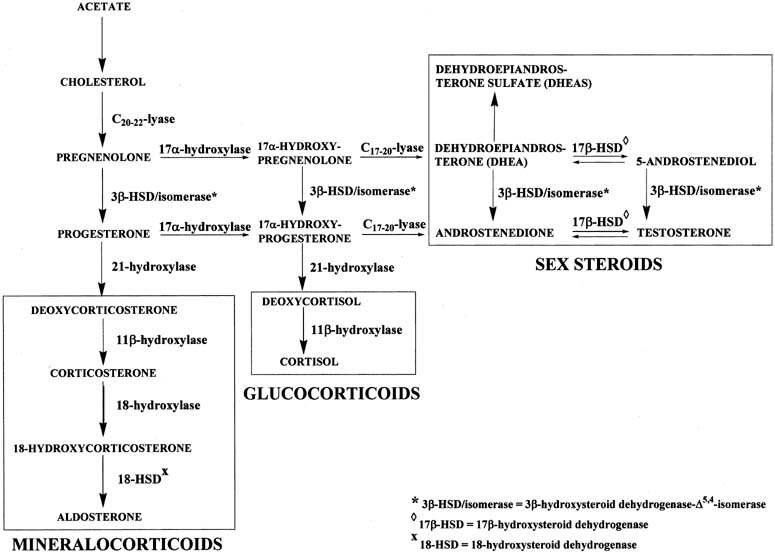
The mineralocorticoid pathway starts with 21-hydroxylation of progesterone to form deoxycorticosterone (DOC). The enzyme in this reaction, 21-hydroxylase, is encoded by the CYP21 gene.11, 12 Deoxycorticosterone is then converted to corticosterone through the action of 11β-hydroxylase. There are two distinct 11β-hydroxylase isoenzymes, both of which are encoded by two genes, CYP11B1 and CYP11B2.13 Corticosterone is hydroxylated at carbon 18 to form 18-hydroxycorticosterone, which is transformed to aldosterone by removal of two hydrogens (oxidation) at carbon 18. These two reactions are catalyzed by 18-hydroxylase and 18-hydroxysteroid dehydrogenase, respectively, which are encoded by the same gene, CYP11B2. Transcription of CYP11B1 is regulated primarily by ACTH, whereas angiotensin II regulates CYP11B2 transcription.14, 15 Similarly, the glucocorticoid pathway begins with 17α-hydroxyprogesterone, which is converted to deoxycortisol and subsequently to cortisol by 21-hydroxylase and 11β-hydroxylase, respectively, in the same manner as the conversion of progesterone to corticosterone. A deficiency of 21-hydroxylase, 11β-hydroxylase, or 3β-HSD in the adrenals may result in congenital adrenal hyperplasia and female pseudohermaphroditism, manifested as a masculinized female fetus.
STEROID HORMONE BIOSYNTHESIS IN THE PLACENTA
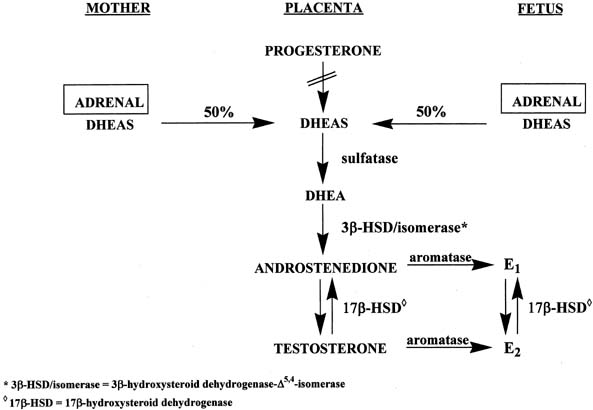
The placenta uses both maternal and fetal, but primarily maternal, cholesterol to form pregnenolone, which is then converted to progesterone by 3β-HSD-Δ4,5-isomerase (see Fig. 6).16 However, the placenta lacks 17α-hydroxylase and C17-20-lyase activities to form androgens and subsequently estrogens. Instead, the placenta uses precursors from the mother and fetus to make estrogens (see Fig. 6).16 Approximately equal amounts of DHEAS originating from the maternal and fetal adrenals enter the placenta, where the sulfate group is split off by sulfatase, yielding DHEA. This androgen is converted to androstenedione and testosterone through the action of 3β-HSD-Δ5,4-isomerase. Subsequently, both androgens are transformed to estrone and estradiol via the enzyme, aromatase.
|
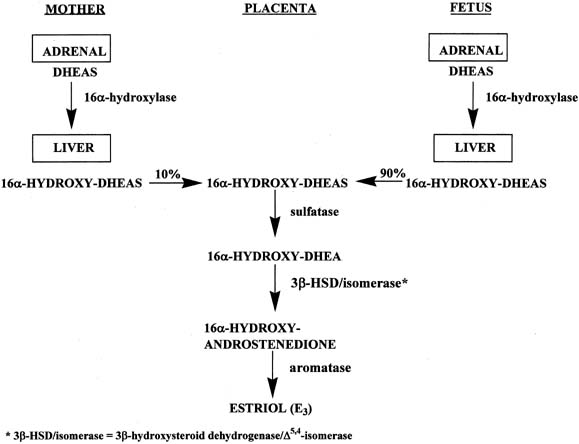
In a manner similar to the formation of estrone and estradiol, placental estriol is formed from 16α-hydroxy-DHEAS, which originates from the fetal and maternal liver (Fig. 7).16 However, in contrast to the precursors of estrone and estradiol, approximately 90% of the 16α-hydroxy-DHEAS entering the placenta is derived from the fetal compartment and only 10% from the mother. Because of the fact that the estriol precursor originates predominantly from the fetus, serum estriol levels have been used for many years to monitor fetal well-being. Use of this marker was replaced with nonhormonal types of antepartum testing.
|
FORMATION OF STEROID HORMONES IN PERIPHERAL TISSUES
So far, the pathways of steroid hormone biosynthesis that have been discussed occur in the endocrine glands. Steroid hormones are also formed in peripheral tissues but not de novo, that is, from acetate or cholesterol. Instead, they are synthesized from circulating precursors made in the endocrine glands. Two important steroidogenic reactions that occur in peripheral tissues are the conversion of androgens to estrogens in adipose tissue, and transformation of testosterone to the more potent androgen, dihydrotestosterone (DHT) in skin.
Adipose tissue has high activity of the enzyme aromatase, which efficiently converts androstenedione to estrone and, to a lesser extent, testosterone to estradiol. This is the mechanism by which estrogens are formed in postmenopausal women.
The conversion of testosterone to DHT occurs via the enzymes, 5α-reductase type 1 and type 2, which are encoded by the SRD5A1 and SRD5A2 genes, respectively.17 The type 1 isoenzyme is found primarily in liver and skin, whereas the type 2 predominates in urogenital tissues. In hyperandrogenic women with idiopathic hirsutism, there is increased activity of 5α-reductase, resulting in increased levels of DHT. Women with this condition have clinical manifestations of hirsutism, acne, and/or alopecia.
TRANSPORT OF STEROID HORMONES
After steroid hormones are secreted into the circulation, they are mostly bound to specific proteins, namely sex hormone-binding globulin (SHBG), corticosteroid-binding globulin (CBG), and/or albumin (Table 2).18 All steroids bind to albumin with low affinity but high capacity. In contrast, SHBG binds with high affinity but low capacity to the sex steroids, including DHT, testosterone, and estradiol. The binding of DHT to SHBG is approximately 3.5-times that of testosterone and eight-times that of estradiol. CBG binds with high affinity but low capacity to corticosteroids, progesterone, and 17-hydroxyprogesterone.
Table 2. Major steroid-binding proteins in blood
| Protein | Steroid(s) Bound | Binding Affinity | Binding Capacity |
| Albumin | All steroids | Low | High |
| α1-Acid glycoprotein (orosomucoid) | Progesterone | Low | High |
| Sex hormone-binding globulin (SHBG) | Dihydrotestosterone, testosterone, estradiol | High | Low |
| Corticosteroid-binding globulin (CBG) | Corticosteroids, progesterone, 17α-hydroxyprogesterone | High | Low |
Table 3 shows the binding distribution of important endogenous steroid hormones in normal women during the menstrual cycle.18 Each steroid is mostly protein-bound; only a small percentage is unbound or free. Free steroids are available for action in target cells and also for metabolism in peripheral tissues. Thus, SHBG and CBG play an important role in regulating the availability of steroid hormones for biologic action and clearance.
Table 3. Binding distribution of principle endogenous steroid hormones in normal women during the menstrual cycle*
| Steroid | SHBG-Bound (%) | CBG-Bound (%) | Albuin-Bound (%) | Unbound (%) |
| Progesterone | 0.63 | 17.40 | 79.5 | 2.37 |
| Estradiol | 37.1 | <0.1 | 60.4 | 1.81 |
| Estrone | 16.2 | <0.1 | 80.1 | 3.58 |
| Testosterone | 65.8 | 2.23 | 30.5 | 1.36 |
| Androstenedione | 6.59 | 1.34 | 84.5 | 7.54 |
| Dihydrotestosterone | 78.2 | 0.12 | 21.1 | 0.47 |
| DHEA | 7.83 | <0.10 | 88.1 | 3.93 |
| Cortisol | 0.18 | 89.60 | 6.4 | 3.83 |
| Corticosterone | 0.22 | 77.80 | 18.6 | 3.33 |
| Aldosterone | 0.23 | 21.60 | 41.3 | 36.9 |
| Deoxycorticosterone | 1.91 | 36.40 | 59.0 | 2.63 |
*Follicular and luteal values were averaged.
(Westphal U. Steroid-Protein Interactions, p 259. Berlin, Springer-Verlag, 1986.)
Of clinical importance is free testosterone, which is often elevated in hyperandrogenic women with clinical manifestations of hirsutism. The free testosterone is regulated by the concentration of SHBG in blood. The higher the SHBG level, the lower the free testosterone level, and vice versa. A number of factors can affect SHBG concentrations in blood. They include obesity, menopause, insulin, and androgens, each of which decreases SHBG levels.19 In contrast, SHBG levels are increased by estrogens, thyroid hormone, liver cirrhosis, and prolonged stress.19
METABOLISM OF STEROID HORMONES
The major sites of steroid inactivation in the body are the liver and, to a lesser extent, the kidney. The inactivation mechanisms include the following: addition of two hydrogens (reduction) to a double bond or ketone group; removal of two hydrogens (oxidation) from a hydroxyl group; addition of a hydroxyl group (hydroxylation) to a carbon in the steroid molecule; and conjugation of steroids by reaction of sulfuric acid or glucuronic acid with a hydroxyl group on the steroid molecule, forming steroid sulfates and glucuronides, respectively.
The process by which steroids are conjugated involves the transformation of lipophilic compounds, which are only sparingly soluble in water, into metabolites that are water-soluble and can readily be eliminated in urine as sulfates or glucuronides. However, steroid glucuronides are excreted more efficiently than steroid sulfates, resulting in much higher concentrations of glucuronidated metabolites in urine, as compared with blood, which contains higher concentrations of the sulfated metabolites. There appears to be a dual mechanism by which this occurs. First, in blood, albumin has a greater affinity for sulfated steroids than for glucuronidated steroids; second, the glomerular filtration rate of the glucuronidated steroids is considerably higher than that of the sulfated compounds.
DYNAMICS OF STEROID HORMONE PRODUCTION AND CLEARANCE
To understand the dynamics of steroid hormone production and clearance, it is essential to define certain parameters that are frequently used to describe the interrelationships of steroid hormones. Quantitation of these parameters is performed by intravenous administration of radioactive steroids to women or men and subsequent measurement of the radioactivity associated with relevant steroids in blood or urine. A description of these techniques and the theoretic aspects used to derive the formulas for quantitation of the different parameters is beyond the scope of this chapter. However, there is an excellent review by Gurpide dealing with the theoretical aspects of the dynamics of hormone production and metabolism.20 For our purposes, four important parameters are defined and the formulas used to quantitate them are described. They include secretion, production rate, metabolic clearance rate, and the transfer constant of conversion.
CALCULATION OF PRODUCTION AND METABOLIC CLEARANCE RATES
The concept of the production rate of a steroid hormone was introduced to describe the rate at which the hormone enters the circulation de novo, regardless of its origin. Therefore, by definition, the production rate of a steroid hormone is equal to the glandular secretion rate of the hormone plus the secretion rates of any other steroids that are converted extraglandularly to the circulating hormone. In the absence of extraglandular sources of the circulating hormone, the production rate of the hormone is identical to its secretion rate. From the practical point of view, the secretion of a steroid hormone by an endocrine gland can be determined by catheterizing the vein draining the organ and demonstrating a higher concentration of the hormone in the venous effluent of the gland than in the peripheral blood. The concentration gradient (difference between the two concentrations) multiplied by the rate of blood flow from the gland yields a rough approximation of the secretion rate.
It has been shown that the physiologic concentration of a steroid hormone in the circulation is directly proportional to its production rate; therefore,
![]()
where PR is the production rate of the hormone, C is its concentration in the circulation, and k is the proportionality constant. This constant was named the metabolic clearance rate (MCR).21
The MCR of a steroid hormone is defined as the volume of blood that is irreversibly cleared of the steroid per unit of time and is usually expressed in liters per day. It is measured by intravenously infusing the radioactive form (usually tritiated) of the steroid, either as a single dose or as a constant rate over a prolonged period (e.g., 2 hours). The radioactive steroid that is infused should have a high specific activity (radioactivity per unit mass), so that only a minute mass of the steroid is administered and the mass does not contribute significantly to the concentration of the endogenous hormone.
The single injection and constant infusion methods yield equivalent MCR for a particular steroid. In the single-dose method, the changes in the concentration of radioactivity (disintegrations per minute [dpm]) associated with the hormone are measured as a function of time. The concentrations of radioactivity are plotted against time, and the areas under the resulting curves are measured. The MCR is then calculated using the following equation: MCR = dose-injected (dpm) divided by area under the radioactivity concentration–time curve (dpm x h/ml).
Because the injected dose is expressed in dpm and the area under the curve as units of dpm per mL multiplied by hours, then the MCR units will be
dpm ÷ (dpm × h)/mL
which can be converted to liters per day. Similarly, if the labeled hormone is infused at a constant rate, a steady state of the radioactive hormone administered will be reached in blood, usually after 1 or 2 hours. A blood sample is taken at this time, and the MCR is calculated using the following equation: MCR = rate of infusion (dpm/h) divided by the concentration of radioactivity associated with the hormone in blood at steady state (dpm/mL).
Because PR ÷ C = MCR, the production rate of a steroid hormone can be readily determined once its MCR and concentration are known. The concentration of the steroid, C, can be measured by radioimmunoassay, whereas the MCR can be determined as described. The following example shows how the production rate of testosterone can be calculated. If the MCR of serum testosterone is found to be 700 L/day in a premenopausal woman and her serum testosterone concentration is measured as 0.35 μg/L, her testosterone production rate can be calculated from the equation, PR = MCR × C. By substituting the values for MCR and C,
PR = 700 L/day × 0.35 μg/L = 245 μg/day
CALCULATION OF INTERCONVERSION OF CIRCULATING STEROIDS
The interconversion rates of circulating steroids are calculated by use of data obtained from experiments in which the radioactive forms of steroids being studied are infused intravenously into a subject at a constant rate. One of the compounds is usually labeled with 3H and the other with 14C. After a certain period of infusion, a steady state is reached for both circulating steroids, and the radioactivity associated with each steroid is measured. From these data, the fraction of circulating compound, for example, androstenedione, that is converted exclusively and irreversibly per unit of time into another compound, such as estradiol, can be calculated from the following formula:
ρAE2 = (3H/14C) E2 isolated/(3H-A/14C-E2) infused
where ρAE2 is the transfer constant of conversion of androstenedione (A) to estradiol (E2); (3H-A/14C-E2) infused represents the ratio of the dpm associated with the mixture of 3H-labeled A and 14C-labeled E2 which is infused; (3H/14C) E2 isolated represents the ratio of the dpm (tritium and carbon 14) associated with estradiol, after its extraction from serum or plasma and purification.
VARIABILITY IN STEROID HORMONE PRODUCTION AND CLEARANCE
It is important to realize that there is a great deal of intersubject and intrasubject variability in the production, circulating levels, and metabolic clearance rates of steroid hormones. In addition, these parameters are affected by episodic fluctuations, diurnal rhythm, phase of the menstrual cycle, and age.
PRODUCTION AND CLEARANCE OF ANDROGENS
The principal androgens in a woman are DHEAS, DHEA, androstenedione, testosterone, and DHT. Of these androgens, DHT is the most potent. It is approximately three times more potent than testosterone. The other androgens have virtually no androgenicity until they are transformed to testosterone or DHT.
Table 4 shows the relative contribution of the adrenals, ovaries, and peripheral tissues to androgen production in premenopausal women.22, 23, 24, 25, 26 Almost all of the DHEA is derived from the adrenals, whereas DHEA comes mostly from the adrenals, but a sizable amount is also produced by the ovaries and peripheral tissues. Approximately equal amounts of androstenedione are derived from the ovaries and adrenals. Approximately 25% of the testosterone production comes equally from the ovaries and adrenals; the remainder comes from peripheral tissues, primarily from androstenedione. Because DHT is not secreted by the endocrine glands, all of it originates from peripheral tissues.
Table 4. Relative contributions (%) of adrenals, ovaries, and peripheral tissues to androgen production in premenopausal women
| Androgen | Adrenals | Ovaries | Peripheral Tissues |
| DHEAS | 90 | 0 | 10 |
| DHEA | 50 | 20 | 30 |
| Androstenedione | 50 | 50 | 0 |
| Testosterone | 25 | 25 | 50 |
| DHT | 0 | 0 | 100 |
Table 5 shows approximate production rates and serum levels of the principal androgens.22, 23, 24, 25, 26 The production rate of DHEAS is highest, followed by DHEA. DHT has the lowest production rate. In postmenopausal women, the production rates are approximately half of those shown for premenopausal women. The production rates of the principal androgens are reflected in the circulating levels of these hormones as shown in Table 5.
Table 5. Production rates and serum levels of the principal androgens in premenopausal women
| Androgen | Production Rate (mg/day) | Serum Levels (ng/mL) Range | |
| Mean | Range | ||
| DHEAS | 11.2 | 3.5–19.6 | 500–2800 |
| DHEA | 6.7 | 4.3–12.6 | 3–8 |
| Androstenedione | 2.8 | 1.4–6.2 | 0.7–3.0 |
| Testosterone | 0.3 | 0.1–0.4 | 0.2–0.7 |
| DHT | 0.04 | 0.02–0.05 | 0.06–0.14 |
The four major circulating androgens derived from the endocrine glands, namely testosterone, androstenedione, DHEA, and DHEAS are excreted in urine almost entirely as 17-ketosteroids. Testosterone is converted extensively to androstenedione. Only a small portion of testosterone produced in the body is metabolized to testosterone glucuronide and is excreted as such in urine. Both androstenedione and DHEA are metabolized primarily to androsterone and etiocholanolone, which are subsequently conjugated as sulfates and glucuronides before their excretion in urine. Although some DHEAS is excreted in urine, DHEA glucuronide is excreted more readily. Urinary 17-ketosteroids consist of conjugated DHEA, androsterone, and etiocholanolone; all have a ketone group at carbon 17. Most of the urinary 17-ketosteroids represent adrenal C19 steroid hormone production and are of no value in assessing ovarian androgen secretion.
There is a wide range in the MCR of the major circulating androgens.27 The MCR of DHEA and androstenedione are approximately 1600 and 2000 L/day in premenopausal women, respectively. In contrast, the MCR of testosterone is 35% and 65% lower than that of DHEA and androstenedione, respectively, because of the high affinity of testosterone for SHBG. Similarly, DHEAS has an extremely low MCR (<10 L/day) because of its high affinity for albumin.
PRODUCTION AND CLEARANCE OF ESTROGENS
There are two principal biologically active estrogens, namely estradiol and estrone. Estradiol is the most potent estrogen in the body and is approximately seven times more potent than estrone. These two estrogens, together with estriol, comprise the three classical estrogens. Estriol has little estrogenic activity.
As shown in Table 6, in a premenopausal woman, approximately 95% of the total estradiol production comes from the ovary, whereas only approximately 40% of the estrone production has an ovarian source; the remaining portions come from peripheral tissues, for example, adipose tissue, using primarily androstenedione and, to a lesser extent, testosterone as substrate. In postmenopausal women, all of the estrogen production is derived from peripheral sources, primarily adipose tissue.
Table 6. Sources of estradiol and estrone in premenopausal and postmenopausal women
| Estrogen | Stage of Life | % of Total Production | |
| Ovarian | Peripheral | ||
| Estradiol | Premenopause | 95 | 5 |
| Postmenopause | 0 | 100 | |
| Estrone | Premenopause | 40 | 60 |
| Postmenopause | 0 | 100 | |
The production rates of estradiol and estrone vary widely during the menstrual cycle (Table 7).28 The rates are lowest in the follicular phase, highest in the preovulatory phase, and intermediate in the luteal phase. As expected, the production rates of these estrogens are very low in postmenopausal women.
Table 7. Production rates of estradiol and estrone in premenopausal and postmenopausal women
| Estrogen | Stage of Life | Phase of Cycle | Production Rate (μg/day) | |
| Mean | Range | |||
| Estradiol | Premenopause | Follicular | 65 | 26–130 |
| Preovulatory | 320 | 200–780 | ||
| Luteal | 160 | 100–390 | ||
| Postmenopause | 13 | |||
| Estrone | Premenopause | Follicular | 110 | 44–220 |
| Preovulatory | 270 | 160–660 | ||
| Luteal | 150 | 88–330 | ||
| Postmenopause | 48 | |||
The different production rates of estradiol and estrone are reflected in the circulating levels of these estrogens (Table 8). Circulating levels of estradiol and estrone average approximately 50 pg/mL in the early part of the follicular phase. As the follicles grow and mature, the levels increase and, in the preovulatory phase, the levels of estradiol and estrone are in the range of 150–600 pg/mL. In the luteal phase, these levels decrease to approximately half of those observed during the preovulatory phase. In contrast, in postmenopausal women, serum estradiol and estrone levels are, on the average, 15 and 40 pg/mL, respectively.
Table 8. Serum levels of estradiol and estrone in premenopausal and postmenopausal women
| Estrogen | Stage of Life | Phase of Cycle | Serum Levels (pg/mL) | |
| Mean | Range | |||
| Estradiol | Premenopause | Follicular | 65 | 20–100 |
| Preovulatory | 250 | 150–600 | ||
| Luteal | 125 | 73–300 | ||
| Postmenopause | 15 | 5–30 | ||
| Estrone | Premenopause | Follicular | 50 | 20–100 |
| Preovulatory | 125 | 75–300 | ||
| Luteal | 70 | 40–150 | ||
| Postmenopause | 40 | 15–55 | ||
Major reactions involved in the metabolism of estradiol and estrone include the following: oxidation of the hydroxyl group or reduction of the ketone group at carbon 17; hydroxylation at carbons 2, 4, 6, 7, 14, 15, or 16; methylation of the hydroxyl group at carbon 2; and conjugation (formation of sulfate or glucuronide) of a hydroxyl group on the estrogen molecule. In urine, the most abundant metabolites of estrone and estradiol are conjugates of 2-hydroxylated and 16α-hydroxylated estrogens.
Estrogens that contain a hydroxyl group on adjacent carbons, for example, 2-hydroxyestrone, 2-hydroxyestradiol, 4-hydroxyestrone, and 4-hydroxyestradiol, are referred to as catechol estrogens. These estrogens compete strongly for the enzyme catechol-0-methyltransferase, which inactivates the catecholamines, dopamine, and norepinephrine. Thus, catechol estrogens may regulate hormonal actions of catecholamines by their inhibitory effect on enzymatic methylation of catecholamines.
Quantitatively, the most important circulating estrogen in women is estrone sulfate. However, it has no inherent biologic activity. During the follicular phase, it is present in amounts as high as 1 ng/mL, and in the luteal phase this amount is almost doubled. Essentially, all of the estrone sulfate production can be accounted for by peripheral formation from estradiol or estrone.
In premenopausal women, the MCR of estrone (1360 L/day per m2) is considerably higher than that of estradiol (790 L/day per m2).28 This is because of the relatively high affinity of SHBG for estradiol. Estrone binds weakly to SHBG.
MEASUREMENT OF STEROID HORMONES
As the term immunoassay implies, the immunoassay method involves an antigen–antibody reaction, where the antigen is the hormone to be measured and the antibody is against this hormone. There are two types of immunoassay methods: one uses an excess of antigen and a limited amount of antibody; the other uses excess antibody. For quantification purposes, each type of immunoassay can use either a radioactive marker (e.g., iodine-125) or a nonradioactive marker, which is usually chemiluminescent, fluorescent, or enzymatic. In every antigen-excess assay or antibody-excess assay used to measure an analyte, there are three components: the standard curve, serum or plasma specimens, and quality-control samples. The assays are performed manually or on an analyzer.
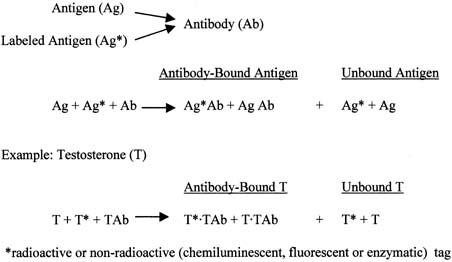
The principle of an antigen-excess type of immunoassay involves competition between the antigen, which is the analyte (e.g., steroid) to be measured, and the labeled form of the same antigen for the active site of the antibody. This is shown more clearly in Figure 8, which depicts the antigen competing with the labeled antigen for the antibody. When all three components are combined, the net result is a mixture of labeled antigen bound to antibody, unlabeled antigen bound to antibody, and unbound labeled and unlabeled antigen. If we use testosterone as an example of the antigen, the net result is a mixture of labeled testosterone bound to the testosterone antibody, unlabeled testosterone bound to the testosterone antibody, and unbound labeled and unlabeled testosterone. From the practical standpoint, nothing happens in a test tube containing these reagents unless one separates antibody-bound from unbound testosterone. Then, by using different concentrations of testosterone standard, one can determine the corresponding amounts of labeled testosterone that are bound to the antibody, and a standard curve can be generated, as shown in Figure 9. Separation of bound and unbound antibody is achieved by one of a variety of different methods, including use of a second antibody (prepared against the first antibody) when an iodinated steroid is used as the labeled antigen, or magnetic particles when a nonradioisotopic marker is used. In an antigen-excess immunoassay, the standard curve shows an inverse relationship between the different amounts of antibody-bound labeled antigen (y-axis) and the different concentrations of the standard (x-axis).
|
|
|
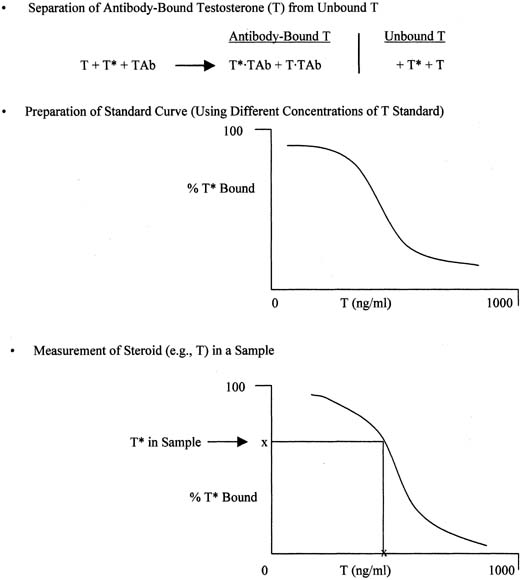
Measurement of a steroid such as testosterone in a serum or plasma sample is obtained by determining the percentage of labeled testosterone that is antibody-bound in the sample and extrapolating the testosterone concentration off the testosterone standard curve, as shown in Figure 9. An appropriate computer program is used to generate the data rapidly.
One of the most widely used antibody-excess immunoassay methods in laboratories is the enzyme-linked immunosorbent assay (ELISA). It is often used to measure proteins. The ELISA method involves addition of an antigen to an excess of antibody, which is attached to a solid phase such as a plastic tube or plate (Fig. 10). After allowing time for the antigen–antibody complex to form, an antibody–enzyme conjugate is added. The antibody in the conjugate is directed against an antigenic site on the antigen, different from that recognized by the first antibody. The result is a “sandwich” type of complex. After addition of the appropriate substrate for the enzyme, the resulting product is measured spectrophotometrically. A linear relationship is found when the optical densities obtained with the different concentrations of the standard are plotted against the concentrations of the standard, in contrast to the curve obtained in the antigen-excess type of assay. The principle of the immunoradiometric assay (IRMA) is similar to that of the ELISA method, except that the initial antibody, which is added in excess, is labeled with a radioactive marker, e.g., iodine-125.
|
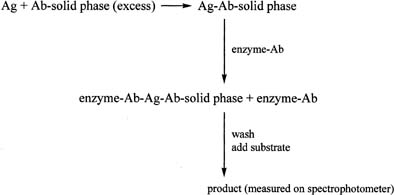
Before use of any immunoassay method for measurement of an analyte in serum or plasma specimens, the assay must be validated with respect to sensitivity, accuracy, precision, and specificity.29 The reliability of each assay is based on the results of the quality-control samples (usually low, medium, and high concentrations) placed at the beginning and at the end of each assay.
REFERENCES
Stocco DM, Clark BJ: The role of the steroidogenic acute regulatory protein in steroidogenesis. Steroids 62:29-36, 1997 |
|
Papadopoulos V, Amri H, Boujrad N, et al: Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids 62:21-28, 1997 |
|
Chung B-C, Picado-Leonard J, Haniu M: Cytochrome P450c17 (steroid 17α-hydroxylase/17,20 lyase): Cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc Natl Acad Sci USA 84:407-411, 1987 |
|
Kagimoto M, Winter JS, Kagimoto K, et al: Structural characterization of normal and mutant human steroid 17-alpha-hydroxylase genes: Molecular basis of one example of combiend 17-alpha-hydroxylase/17,20 lyase deficiency. Mol Endocrinol 2:564-570, 1988 |
|
Rhéaume E, Lachance Y, Zhao H-F, et al: Structure and expression of a new complementary DNA encoding the almost exclusive 3β-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase in human adrenals and gonads. Mol Endocrinol 5:1147-1157, 1991 |
|
Lorence MC, Corbin CJ, Kamimura N, et al: Structural analysis of the gene encoding human 3 beta-hydroxysteroid dehydrogenase/delta 5-4-isomerase. Mol Endocrinol 4:1850-1855, 1990 |
|
Lachance Y, Luu-The V, Verreault H, et al: Structure of the human type II 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4-isomerase (3 beta-HSD) gene: Aadrenal and gonadal specificity. DNA Cell Biol 10:701-711, 1991 |
|
Andersson S, Moghrabi N: Physiology and molecular genetics of 17β-hydroxysteroid dehydrogenases. Steroids 62:143-147, 1997 |
|
Geissler WM, Davis DL, Wu L, et al: Male pseudohermaphroditism caused by mutations of testicular 17β-hydroxysteroid dehydrogenase 3. Nature Genet 7:34-39, 1994 |
|
Simpson ER, Mahendroo MS, Means GD, et al: Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endo Rev 15:342-355, 1994 |
|
Carroll MC, Campbell RD, Porter RR: Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc Natl Acad Sci USA 82:521-525, 1985 |
|
White PC, Grossberger D, Onufer BJ, et al: Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA 82:1089-1093, 1985 |
|
Mornet E, Dupont J, Vitek A, et al: Characterization of two genes encoding human steroid 11 beta-hydroxylase (P-450(11) beta). J Biol Chem 264:20961-20967, 1989 |
|
Lifton RP, Dluhy RG, Powers M, et al: Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat Genet 2:66, 1992 |
|
Pascoe L, Curnow KM, Slutsker L, et al: Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP1lB1 and CYP1lB2. Proc Natl Acad Sci USA 89:8327-8331, 1992 |
|
Diczfalusy E: Steroid metabolism in the feto-placental unit. In: Pencile A, Finzi C (eds): The Feto-Placental Unit, p 65. Amsterdam, Excerpta Medica, 1969 |
|
Russell DW, Berman DM, Bryan JT, et al: The molecular genetics of steroid 5α-reductases. Recent Prog Horm Res 49:275-284, 1994 |
|
Westphal U: Steroid–Protein Interactions. Berlin, Springer-Verlag, 1986 |
|
Rosner W: Plasma steroid-binding proteins. Endocrinol Metab Clin North Am 20:697-720, 1991 |
|
Gurpide EL: Tracer Methods in Hormone Research. New York, Springer-Verlag, 1974 |
|
Tait JF, Burstein S: In vivo studies of steroid dynamics in man. In: Pincus G, Thimann KV, Astwood EB (eds): The Hormones, p 441, Vol 5. New York, Academic, 1964 |
|
Abraham GE: Ovarian and adrenal contribution to peripheral androgen during the menstrual cycle. J Clin Endocrinol Metab 39:340-346, 1974 |
|
Longcope C: Adrenal and gonadal androgen secretion in normal females. Clin Endocrinol Metab 15:213-228, 1986 |
|
Vermeulen A: Plasma androgens in women. J Reprod Med 43:(S):725-733, 1998 |
|
Adashi EY: The climacteric ovary: A viable endocrine organ. Sem Reprod Endocrinol 9:200-205, 1991 |
|
Davis SR: Androgens. In: Lobo RA, Kelsey J, Marcus R (eds): Menopause Biology and Pathobiology, pp 445–448. San Diego, Academic Press, 2000 |
|
Longcope C: Androgen metabolism and clearance. In: Azziz R, Nestler JE, Dewailly D (eds): Androgen Excess Disorders in Women. Philadelphia, Lippincott-Raven Publishers, 1997 |
|
Baird D, Horton R, Longcope C: Steroid dynamics under steady state conditions. Recent Prog Hormone Res 25:611, 1969 |
|
Nakamura RM, Stanczyk FZ: Immunoassays. In Lobo RA, Mishell DR Jr, Paulson RJ, et al (eds): Mishell's Textbook of Infertility, Contraception and Reproductive Endocrinology, pp 76–88. 4th ed.. Malden, MA Blackwell Science, 1997 |