Gonadotropin-releasing Hormone (GnRH) and the GnRH Receptor (GnRHR)
Authors
INTRODUCTION
Normal reproductive function requires the precise temporal and quantitative regulation of hormone secretion at all levels of the hypothalamic–pituitary–gonadal axis. The hypothalamus contains gonadotropin-releasing hormone (GnRH) neurons which secrete pulsatile GnRH into the hypophyseal portal blood system through which it is transported to the anterior pituitary gland. GnRH binds to its receptor on gonadotrope cells, stimulating the biosynthesis and secretion of the gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH and FSH travel through the peripheral circulation, acting at the gonads to stimulate gametogenesis (i.e., the development of mature eggs and sperm) and steroidogenesis (i.e., synthesis of the gonadal hormones ‒ estrogen, progesterone, and androgens). In the majority of physiologic conditions, the gonadal steroids feedback at the hypothalamus and pituitary to decrease GnRH and gonadotropin secretion. An exception is at the time of the periovulatory LH surge in females, believed to be due to positive feedback by rapidly rising estradiol levels.
Isolated in the early 1970s, GnRH was one of the earliest of the hypothalamic-releasing hormones to be sequenced and characterized.1 It was initially believed that separate hypothalamic factors were responsible for LH and FSH secretion and, as a result, GnRH was originally termed luteinizing hormone-releasing factor (LHRH). This latter term is still occasionally used in the literature.
GnRH consists of 10 amino acids which have been highly conserved over millions of years of evolution (Figure 1). In common with other neuropeptides, GnRH is synthesized as part of a large prohormone that is cleaved enzymatically and further modified within the secretory granules. This 92 amino acid precursor protein is proteolytically cleaved to generate: (1) the GnRH decapeptide, (2) a 23 amino acid signaling sequence which directs intracellular packaging and secretion, (3) a three amino acid (Gly-Lys-Arg) proteolytic processing site, and (4) a 56 amino acid GnRH-associated protein (GAP) that is secreted with GnRH. The function of GAP is unknown but it has been proposed to inhibit prolactin secretion in some species.2
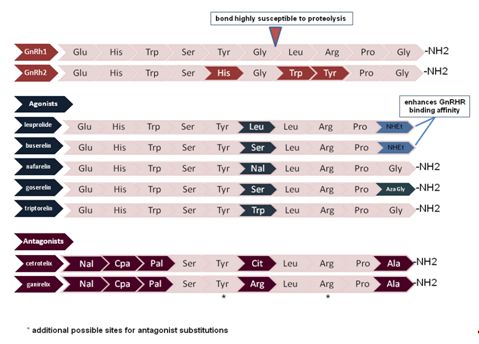 Fig. 1 Comparison of the amino acid compositions of GnRH1 and GnRH2 with commonly used agonists and antagonists. Amino acid variations as compared with GnRH1 are color coded. Nal, napthyl-alanine; Aza-Gly, Aza-glycine (alpha carbon replaced with a nitrogen); CpA, cyanoproprionic amino acid; Pal, pyridyl-alanine
Fig. 1 Comparison of the amino acid compositions of GnRH1 and GnRH2 with commonly used agonists and antagonists. Amino acid variations as compared with GnRH1 are color coded. Nal, napthyl-alanine; Aza-Gly, Aza-glycine (alpha carbon replaced with a nitrogen); CpA, cyanoproprionic amino acid; Pal, pyridyl-alanine
GnRH has a short half-life of approximately 2–4 min due to rapid cleavage by peptidases. As a result of this rapid degradation as well as massive dilution, the peripheral circulation does not contain biologically active concentrations of GnRH. Serum LH and FSH levels are used clinically as surrogate markers for the presence of pulsatile GnRH secretion. LH is a more accurate indicator of GnRH pulse characteristics (i.e., frequency and amplitude) than is FSH which has a longer half-life.
The majority of our understanding of GnRH and GnRH-receptor (GnRHR) function is based on studies of a single isoform of each; however, recent studies have identified additional forms as described in subsequent sections. For this review, the terms "GnRH" and "GnRHR" will refer to the type 1 GnRH and GnRHR, respectively. Note also that human gonadotropin-releasing hormone should be abbreviated with all capitals (i.e., GNRH) according to the new nomenclature. As much of the available data have been obtained in non-human species, we have chosen to use the more common abbreviations.
MIGRATION OF GnRH NEURONS DURING DEVELOPMENT
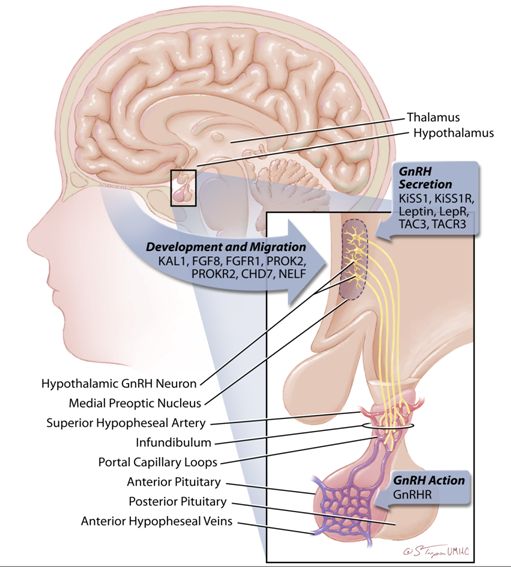
While the majority of neural cells arise from neurons within the developing nervous system itself, GnRH neurons are unusual in that they are derived from progenitor cells in the epithelium of the olfactory placode. These nascent GnRH neurons migrate along the vomeronasal axons, across the cribiform plate and into the mediobasal hypothalamus where migration ceases and the neurons detach from their axonal guides.3 Patients with delayed puberty due to abnormal GnRH neuronal migration frequently have associated anosmia, or the inability to smell, which reflects the fact that the GnRH neurons share common embryonic origins and migratory pathways with olfactory neurons. The identification of genes which direct normal GnRH migration and function is an active area of research. A long list of soluble factors have been identified which appear to be critical for the ultimate development of a network which contains the appropriate number and location of GnRH neurons. These include pathway markers (netrin‑1), cell cycle arrest (Gas6), signaling molecules (GABA), growth factors (fibroblast growth factors), and adhesion molecules (tenascin, phosphacan, and laminin).4 Mutations in these genes result in clinical phenotypes including pubertal delay and infertility, and are discussed in the Section The GnRH receptor (GnRHR) below.
Estimates of the number of GnRH neurons vary, but are in the range of a few thousand, a remarkably small number in view of their critical function.5 The cell bodies of these neurons are scattered across a number of hypothalamic nuclei, with the majority residing in the arcuate nucleus of the medial basal hypothalamus in the human. Most GnRH neurons send axonal projections to the median eminence which abuts the hypothalamic–pituitary portal system. This system consists of capillaries that arise from the superior hypophyseal arteries, traverse the pituitary stalk, and then form a capillary network within the pituitary gland. This anatomic relationship allows minute quantities of GnRH secreted by these axonal terminals to have direct access to the pituitary gonadotropes. The primary direction of this hypophyseal portal system is from the hypothalamus to the pituitary; however, retrograde flow also exists and provides a short feedback loop from the pituitary back to the hypothalamus.
A subset of GnRH neurons extend axons to other portions of the CNS, including the limbic system. While these projections are not directly involved in the modulation of gonadotropin secretion, they may help to link hormonal status to reproductive behavior.6 Thus, GnRH neurons are positioned to both receive and generate neural and hormonal inputs, allowing for the complex integration of reproductive function and broader physiologic status.
GnRH SECRETION FROM THE HYPOTHALAMUS
Requirement for pulsatile GnRH secretion
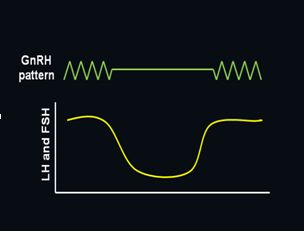
In an elegant series of experiments, Ernst Knobil and colleagues demonstrated that pulsatile GnRH is required to achieve sustained gonadotropin secretion.7 Using a primate model, continuous infusion of GnRH was found to rapidly suppress both LH and FSH secretion, an effect that was readily reversed with a return to pulsatile stimulation (Figure 2). Loss of the GnRH response with continuous treatment is now known to be due to rapid uncoupling of the GnRH receptor from its intracellular signaling molecules followed by downregulation of receptor number. This characteristic is exploited clinically by administration of long-acting GnRH agonists to treat steroid-dependent conditions such as endometriosis, leiomyomas, breast cancer, and prostate cancer.8 Pulsatile activity is currently believed to be an intrinsic property of GnRH neurons with hormonal and neural inputs providing modulatory effects; however, it has never been definitively proven whether the pulse generator is within a single GnRH neuron or a property of the neuronal network.9, 10
{kind=link}
GnRH neuronal activity varies across the lifespan as is reflected by changes in gonadotropin levels and, ultimately, gonadal steroid and gamete production. In the human, GnRH is detectable in the hypothalamus by 10 weeks gestational age with FSH and LH produced by 10–13 weeks when the vascular connection between the hypothalamus and pituitary gland has developed. Gonadotropin levels peak at mid-gestation and then decline towards term due to negative feedback at both the hypothalamus and pituitary by the high levels of placental steroids. With the withdrawal of placental steroids at birth, gonadotropins rise and remain elevated for the first 1–2 years in girls and first 6 months in boys with a subsequent decrease for the remainder of childhood. The etiology for this suppression is unknown but may involve inhibition by the neurotransmitters g-aminobutyric acid (GABA) and neuropeptide Y (NPY).11, 12
{kind=link}
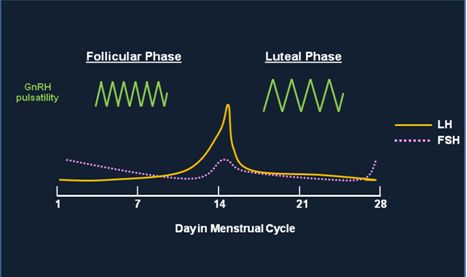
Fig. 3 This figure shows the change in frequency of GnRH pulsatile secretion throughout the menstrual cycle and how the change affects the secretion of both LH and FSH. GnRH is secreted at a higher frequency pulse rate during the first part of the menstrual cycle leading to a preferential secretion of LH and the LH surge which is noted on Day 14. During the second half of the menstrual cycle, GnRH is secreted at a lower frequency pulse rate which causes a preferential secretion of FSH. The amplitude of GnRH pulses increases in the luteal phase; the effects of amplitude changes on LH/FSH secretion are less understood than changes in pulse frequency
GnRH pulse characteristics change once again at menopause. There has been longstanding consensus that women are born with the full cohort of follicles that they will have during their lifetime. With depletion of follicular number, estrogen levels decrease with subsequent loss of negative feedback and resultant increases in GnRH secretion. GnRH pulses occur approximately every 50–55 min in younger postmenopausal women which is comparable to a normally cycling woman in the late follicular phase and midcycle surge. As women age from the 5th to the 8th decade, the frequency of GnRH pulses decreases by around 35%.17 Of interest, data are emerging which suggest that stem cells may exist in the ovary; however, this is currently a controversial area of investigation.18, 19
Hormonal regulation of GnRH secretion
Estradiol likely acts at both the hypothalamus and pituitary gland to exert negative and positive feedback effects on GnRH secretion and gonadotropin release. Within the hypothalamus, altered GnRH pulsatility may be a result of direct or indirect effects on the GnRH neurons. For many years, it was believed that GnRH neurons lacked estrogen receptor expression, suggesting that all effects on these neurons were achieved via connecting interneurons. However, more recent studies have demonstrated that the estrogen receptor, ERb, is expressed by at least a subset of GnRH neurons. The presence of ERa is more controversial, although ERa mRNA has been clearly identified in some studies.20, 21 Some of these discrepancies may be attributable to species differences. There is also substantial evidence to support a direct negative effect by estrogens at the pituitary level.22 For example, studies have been performed in women with GnRH deficiency who were treated with pulsatile GnRH followed by estradiol. Estradiol was able to blunt GnRH-mediated increases in gonadotropin expression, despite a lack of potential feedback at the hypothalamus.23
Circulating estradiol levels are a reflection of the degree of ovarian follicular development. Although low estradiol levels feedback negatively as just described, rapidly increasing estradiol levels exert a positive feedback effect and are responsible for generating the pre-ovulatory gonadotropin surge. It has been demonstrated in a monkey model that estradiol levels of 200-400 pg/ml persisting for at least 36 hours are adequate to generate an LH surge.24
In the normal physiological situation, estradiol likely acts via both the hypothalamus and pituitary to trigger the LH surge. Within the pituitary, estradiol and GnRH increase GnRHR expression, resulting in increased pituitary sensitivity to GnRH pulses. Hypothalamic GnRH secretion is also increased at the time of the surge, as directly measured in sheep and rats; however, this change may not be a required. It is possible to generate an estradiol-induced surge experimentally despite holding GnRH pulse frequency and amplitude constant in animals lacking endogenous GnRH activity. Thus, even though GnRH release normally increases at the time of the surge, this increase appears to be facilitory rather than essential for production of a surge.25, 26, 27
Progesterone also decreases GnRH secretion at the level of the hypothalamus. It is controversial whether GnRH neurons express progesterone receptors and, therefore, interneurons expressing these receptors may be responsible for feedback effects. It has been clearly established that estrogen priming is necessary to observe a progestational effect, undoubtedly due to the marked ability of estrogen to upregulate progesterone receptor expression.28, 29
A number of neurotransmitters and neuropeptides are believed to act as intermediaries between circulating gonadal steroid levels and GnRH pulse secretion. For example, estrogen promotes endorphin secretion with a further increase in the presence of progesterone. Endorphins, along with other opioids, suppress hypothalamic GnRH release. Thus, endorphin levels peak with the high steroid levels found in the mid-luteal phase, suggesting that opioid tone may act with progesterone to decrease GnRH pulse frequency in this phase relative to the follicular phase. NPY, norepinephrine and dopamine-secreting neurons are also likely to be important for modulation of GnRH neuronal activity. In addition, it has been demonstrated that CRH inhibits hypothalamic GnRH secretion, both directly and by augmenting endogenous opioid secretion. Women with hypothalamic amenorrhea and women under high levels of stress experience hypercortisolism, suggesting that this may be at least one pathway by which these pathophysiologic states interrupt reproductive function.30, 31
Kisspeptin activation of GnRH neurons
The discovery of the kisspeptin neuronal system has substantially advanced our understanding of the regulation of the hypothalamic GnRH system at puberty, across the female reproductive cycle, and at menopause. The majority of the kisspeptin studies have been performed in rodents, but at least fundamental similarities are being documented in primate models. Humans with mutations in the kisspeptin receptor experience hypogonadotropic hypogonadism, strongly suggesting a key role in GnRH neuronal function in humans.
Kisspeptin is a 145 amino acid polypeptide which binds to a specific receptor, termed the G protein-coupled receptor 54 (GPR54). Kisspeptin secreting neurons are located in the arcuate and anteroventral periventricular (AVPV) nuclei of the hypothalamus. The majority of kisspeptin neuronal mapping has been done in rodents, but the presence of kisspeptin secreting neurons in the arcuate nucleus has been established in humans.32
Kisspeptin neurons may act directly or transsynaptically by way of neuorotransmitters.33 In the rodent model, neurons from both the arcuate and AVPV nuclei send projections to the medial preoptic area of the hypothalamus where the GnRH neurons reside. Arcuate kisspeptin neurons are not as abundant as AVPV neurons, but are in much closer proximity to GnRH neurons in the median eminence. Approximately 50–75% of GnRH neurons express the kisspeptin receptor.34 Kisspeptin acts primarily at the level of the hypothalamus to stimulate GnRH secretion.35 Nevertheless, a number of studies have demonstrated the ability of kisspeptins to act directly on pituitary gonadotrope cells to stimulate LH release.36, 37, 38
The onset of puberty is marked by an increase in synaptic connections between Kiss1 and GnRH neurons. Evidence also suggests an overall elevation in kisspeptin tone as well as enhanced kisspeptin signaling efficacy. These changes appear to occur primarily via the AVPV neurons and, in the female, may be triggered by subtle increases in ovarian estrogen production.39, 40
Expression of the KISS1 gene is under the control of both estrogens and androgens.41 Kisspeptin neurons in the arcuate nucleus are believed to be involved in negative feedback by gonadal steroids, while the AVPV neurons are likely involved in the pre-ovulatory surge of LH and FSH through positive feedback by estradiol.42 Stated differently, estrogen exerts opposite feedback effects on the two populations of kisspeptin neurons. Sexual dimorphism also exists in that estrogen is unable to generate a surge in the male, possibly due to the greater number of kisspeptin neurons in the AVPV of adult females compared to males.43
Kisspeptin expression is further modified by the adipose-derived factor, leptin. A 167 amino acid polypeptide, leptin is secreted by white adipose tissue and regulates body weight by decreasing food intake and increasing energy expenditure. Within the arcuate nucleus, leptin has been shown to increase activity of the kisspeptin neurons. Conversely, decreased expression of KISS1 mRNA can be seen with hypoleptinemia. This interaction between leptin and the kisspeptin system may be one mechanism by which metabolic status is linked to reproductive function, for example in periods of starvation/energy-deprivation as occurs with eating disorders or excessive exercise.44, 45
GnRH isoforms
In the past decade, it has become clear that a second form of GnRH is expressed in addition to the "classic" type 1 GnRH (GnRH1) (Figure 1). This alternate form, called GnRH2, differs from GnRH1 by 3 amino acids at positions 5, 7, and 8. A third form of GnRH, GnRH3, has been reported in several fish species but is not currently thought to be present in humans. In the human, the gene encoding GnRH1 is found on chromosome 8p11, whereas the gene for GnRH2 is found on chromosome 20p13. The genes for both forms of GnRH are composed of four exons and three introns which encode an identically organized precursor polypeptide. Expression of the GnRH1 and GnRH2 genes are driven by different promoter sequences, suggesting that their transcriptional regulation likely differs. Detailed analysis of the GnRH1 gene promoter sequence has identified a number of DNA-regulatory regions which provide for tissue-specific expression.46, 47
GnRH1 and GnRH2 are expressed in overlapping tissue patterns; however, as a general rule, GnRH2 can be detected in a broader range of tissues and is present at higher levels outside of the brain.48 In addition to hypothalamic sources, GnRH1 immunoreactivity has been detected within the pituitary gland itself. The presence of GnRH1 in human thyrotropes and somatotropes, suggests additional roles for GnRH signaling in the pituitary. GnRH1 and GnRH2 have been found in reproductive tissues including the ovary, prostate, endometrium, breast and placenta as well as in tumors derived from these tissues.49 GnRH2 is particularly abundant in the prostate, kidney and bone marrow, consistent with the view that GnRH2 has a wide range of reproductive and nonreproductive functions. In the central nervous system, both isoforms have been localized to the preoptic and mediobasal regions with additional GnRH2 expression in midbrain regions such as the hippocampus, caudate nucleus, and amygdala. Unlike GnRH1 neurons, GnRH2 neurons are not derived from the olfactory placode. Interestingly, GnRH2 is also expressed at the median eminence and preferentially stimulates FSH secretion in the sheep, suggesting that GnRH2 may modulate GnRH1 effects on gonadotropes.50, 51, 52, 53
Studies have demonstrated that GnRH1 and GnRH2 are differentially regulated by gonadal steroids. As shown in human granulosa-luteal cells, estradiol decreases GnRH1 and increases GnRH2 mRNA levels in a dose- and time-dependent manner. Furthermore, in these same cells, treatment with the progesterone antagonist RU486 increases GnRH2 transcript number but does not alter GnRH1 expression.54 As both GnRH1 and GnRH2 coexist in the ovary, divergent regulation may be one mechanism by which these GnRH isoforms generate different functional effects in this tissue.
THE GnRH RECEPTOR (GnRHR)
GnRH receptor tissue expression and function
The gene which encodes the GnRHR is located on chromosome 4 in the human. While additional GnRHR isoforms have been identified in other species, at present, only a single GnRHR peptide has been definitively detected in humans. Although the mRNA for a putative type 2 receptor has been detected in endometrium, ovary, testes, placenta, and prostate cells, this transcript contains a frame-shift that generates a premature stop codon and lacks a methionine translation initiation codon. As such, it is unlikely that this receptor is expressed in a functional form. Nevertheless, it has been proposed that the truncated human GnRHR2 may play a modulatory role in GnRHR1 expression by perturbing normal processing of the latter.55
As previously described, GnRH is expressed in a wide range of non-pituitary tissues. Likewise, GnRH receptors have been identified in an extensive array of normal tissues, as well as benign and malignant tumors derived from these tissues. GnRHR expression has been detected in reproductive tissues including the ovary, testes, endometrium, myometrium, prostate, breast and placenta.56 Unique transcription initiation sites have been characterized in pituitary, ovarian, and placental tissues which likely explain tissue-specific expression of this transcript. Co-localization of GnRH and its receptor in multiple cell types strongly suggests that GnRH may act in an autocrine/paracrine manner beyond the regulation of gonadotropin secretion.57, 58, 59, 60, 61
Within reproductive tissues, GnRH and its receptor are thought to play a role in normal breast and ovarian development. In the adult ovary, the expression of GnRHR correlates with stage of follicular growth, with high levels of GnRHR binding in granulosa cells from late follicles and developing corpora luteal cells, but limited binding in primordial, early antral, and preovulatory follicles. This stage-specific timing suggests significant effects on cellular proliferation, differentiation, and steroidogenesis.62, 63 GnRH has been implicated in the regulation of sperm motility and sperm–oocyte interactions, growth inhibition in reproductive tumors, and human chorionic gonadotropin release in the placenta. In non-reproductive tissues, GnRH has been reported to modulate neuronal migration, visual processing, digestive tract function, and immune T cell chemotaxis. Studies in endometrial, ovarian, and prostate tumor cell lines have implicated GnRH in mediating cell growth, angiogenesis, invasion, and metastasis.64
Intracellular signaling
The GnRH receptor (GnRHR) is a member of the rhodopsin-like G protein-coupled receptor superfamily characterized by the presence of a hydrophilic extracellular domain, an intracellular domain, and a hydrophobic transmembrane domain that spans the cell membrane seven times.65 When bound to hormone, these receptors undergo a conformational change, activate intracellular signaling pathways and, through a series of phosphorylation events, ultimately modulate transcription of multiple genes within the target cell. Receptor activation also induces the formation of receptor clusters which are internalized and then either degraded in lysosomes or shuttled back to the cell surface. The GnRHR differs from other G-protein coupled receptors in that it has a relatively short intracellular carboxy terminal tail that slows receptor internalization and prevents rapid desensitization.66, 67
A substantial amount is known about the intracellular pathways that are downstream of GnRH receptors in the pituitary gonadotropes. The GnRHR is coupled to the Gaq/11 member of the G-protein family which links to inositol phospholipid (IP3) turnover, cleavage of PIP2, followed by activation of the protein kinase C (PKC) signaling system and calcium mobilization. Protein kinase C further stimulates the MAP kinase and ERK1/2 pathways. Although less well-appreciated, GnRHR activation also activates the cAMP/protein kinase A (PKA) signaling pathway via both Gas and the calcium/calmodulin system. Activation of these pathways ultimately results in the stimulation of gonadotropin biosynthesis and secretion.68
Available data suggest that activation of the GnRHR may regulate downstream signaling systems in a cell-specific manner. In contrast to effects in the pituitary, activation of GnRH receptors in peripheral tissues has been shown to preferentially activate the Gai pathway which inhibits cAMP production.69 These differences are unlikely to be due to variations in GnRH receptor protein sequence as the GnRHR cDNA has been found to be identical in all tissues studied to date. While it is possible that cell-specific post-translational splicing may occur, it has been proposed that variations in phosphorylation or glycosylation state are more likely explanations for the observed functional differences.
Multiple additional mechanisms have been proposed by which variable cell-specific GnRHR signaling may be achieved. As one possibility, intracellular signaling may also differ based on the identity of the ligand which is bound to the receptor as this will determine conformational changes which will ultimately impact signaling. For example, although cetrorelix is widely regarded to be a competitive antagonist, it has been shown to act as an agonist on peripheral GnRH receptors. As discussed in the section ' GnRH isoforms', GnRH exists as two isoforms in the human. GnRH1 is more effective than GnRH2 in positively responding prostate carcinoma cell lines, while GnRH2 is more effective in negatively responding cell lines.70
Signaling may also be altered by ligand affinity. Although native GnRH has the same affinity for pituitary and placental GnRH receptors, the GnRH agonist buserelin binds to pituitary receptors with over 100-fold higher affinity. GnRH dose and the number of cell-surface GnRH receptors may also impact signaling and, thereby, effects on cellular physiology.71, 72
Hormonal regulation of GnRHR expression
GnRHR expression in the gonadotrope is markedly upregulated by GnRH itself, as well as by estrogen, progesterone, and androgens.73 Of note, the vast majority of studies have been performed in rodent and sheep models and significant species-specific disparities may occur. Cell-specific differences in regulation are also very likely, although currently poorly characterized. Nevertheless, it is widely accepted that pulsatile GnRH is required for GnRHR expression on the gonadotropes with higher pulse frequencies resulting in maximal stimulation. In contrast, continuous GnRH down-regulates GnRHR cell-surface receptor. The effect of continuous GnRH exposure to eliminate the pituitary GnRH response is exploited in the clinical use of GnRH agonists (see section on GnRH agonists and antagonists below). Constant GnRH exposure presumably does not decrease GnRHR expression in peripheral tissues in which local GnRH levels are unlikely to be pulsatile.
Pituitary GnRHR expression is also increased by estradiol. Increased receptor expression and/or stimulation by increased GnRH pulse frequency and estradiol levels facilitate the generation of the peri-ovulatory LH surge. Progesterone has been reported to suppress GnRHR number in the pituitary gland as observed at times of high circulating levels during the female reproductive cycle and pregnancy.74
Chronic stress is known to be associated with reproductive dysfunction, attributable in part to associated increases in adrenally derived glucocorticoids such as cortisol. Cortisol has been reported to disrupt GnRH pulsatility and may also have direct action at the pituitary. Glucocorticoids have been shown to have a direct positive effect on GnRHR transcription of the mouse promoter; however, they inhibit GnRH-induced LH secretion from bovine and porcine pituitary cells.75, 76
Inhibins and activins are closely related peptides, with inhibins consisting of heterodimers of an a-subunit and b-subunit, while activins are b-subunit homodimers. While both may be expressed in the pituitary, the majority of data suggest that inhibins are primarily products of the ovarian granulosa and luteal cells. In contrast, activins are produced in multiple tissues including the pituitary and exert their effects locally. Inhibin has been found to prevent GnRH-mediated stimulation of GnRHR mRNA levels, while activin may play an important role in maintaining basal GnRHR expression in gonadotropes.77
GnRHR promoter activity is also stimulated by the neuropeptide, pituitary adenylate cyclase activating polypeptide (PACAP). Hypothalamic PACAP is released into the portal vasculature, binding to specific PACAP receptors on gonadotrope cell membranes and acting to augment gonadotropin secretion. PACAP is also secreted by gonadotropes and folliculostellate cells in the pituitary and may therefore act as an autocrine–paracrine factor to maintain GnRHR number. In addition, PACAP is expressed in a wide array of reproductive and non-reproductive tissues and therefore may regulate expression of type 2 GnRH receptors.78
GnRH AGONISTS AND ANTAGONISTS
Pharmacology
GnRH itself and the gonadal steroids produced in response to GnRH stimulation play a role in normal reproduction and in an array of pathophysiologic states. In response to this observation, a substantial number of GnRH analogues have been developed for therapeutic use. Naturally occurring GnRH has a half-life of 2–4 min which can largely be accounted for by breakage of the glycine-leucine bond between amnio acids 6 and 7 (Fig. 1). GnRH agonists have a substitution for glycine at position 6 which significantly increases the plasma half-life compared to the native hormone. Some agonists also substitute ethylamide for glycine at position 10 which increases the affinity for GnRHR. GnRH antagonists have amino acid modifications at many more positions including 1, 2, 3, 6, 10, and sometimes 8 and 5.79
Clinical uses
GnRH agonists can be used in pulsatile or continuous regimens to treat estrogen-dependent conditions including endometriosis, uterine leiomyomas, precocious puberty, and menorrhagia. Pulsatile GnRH may be used to induce puberty in patients with hypogonadotropic hypogonadism and has been used successfully to induce follicular development or sperm production with resultant pregnancy.80 GnRH analogues are used extensively during in vitro fertilization cycles to prevent a spontaneous LH surge and allow for retrieval of the mature oocytes.
GnRH agonists are also used extensively for the short term treatment of endometriosis and uterine fibroids, with an average reduction of 30–50% in fibroid volume during the treatment period. Unfortunately, osteoporosis will develop under the low estrogen conditions induced by prolonged GnRH agonist use. Therefore, treatment is generally limited to 6 months unless some form of estrogen add-back regimen is instituted. As a result, GnRH agonists are generally used in preparation for surgery or in the peri-menopausal woman awaiting menopause and the natural cessation of ovarian function.81, 82
With the appreciation that many tissues express GnRH and GnRH receptors, these analogues may be of use for their direct action at the target tissue rather than just indirectly through decreases in gonadal steroid production. For example, GnRH analogues are also of potential use in the treatment of a wide variety of cancers which express GnRHR, including breast, pancreatic, and ovarian cancers.
One of the potential drawbacks of using GnRH agonists is the so called “flare effect.” The flare effect refers to the transient increase in LH and FSH release from stored secretory granules in response to initiation of GnRH agonist treatment prior to the ultimate downregulation of GnRHR expression caused by saturation of the GnRHR. It takes approximately 1–3 weeks of GnRH agonist use to obtain a fully hypogonadotropic hypogonadal state. Side-effects of GnRH agonist therapy are related to sex hormone deficiency and are similar to menopausal symptoms, including hot flushes, vaginal dryness, and osteopenia.83, 84 GnRH antagonists have the benefit of bypassing the flare effect and immediately decreasing gonadotropin levels with a therapeutic effect seen in 24–72 hours. The original GnRH antagonists were of limited clinical utility due to their associated histamine release; however, the newer antagonists do not have this undesirable side-effect. GnRH antagonists have mainly been used to treat infertility and advanced-stage prostate cancer. More patient-friendly, orally active formulations are under development and are being studied for the treatment of a broader array of conditions.85
An emerging use of GnRH analogues is for fertility preservation in patients undergoing chemotherapy for cancer or rheumatologic diseases such as lupus erythematosus. Chemotherapy-induced early menopause has been well-established in the literature and with improved survival rates especially among premenopausal patients, fertility preservation has become increasingly important. A study on premenopausal breast cancer patients, demonstrated a 17% reduction in early menopause in patients receiving the GnRH analogue triptorelin prior to and during chemotherapy.86 An important study is in progress which will examine the efficacy of GnRH analogue treatment in maintaining ovarian function in patients receiving cyclophosphamide as therapy for lupus. It should be noted that the efficacy of GnRH analogue treatment for fertility preservation remains controversial. Where available, oocyte freezing or embryo freezing are preferable approaches at this time.
INHERITED ABNORMALITIES IN GnRH ACTION
Abnormalities in GnRH stimulation of pituitary gonadotropin secretion may present with varying degrees of hypogonadotropic hypogonadism ranging from total absence of gonadal steroid production and lack of pubertal development to the delay of puberty to infertility due to inadequate stimulation of oocyte or sperm production. Patients with hypogonadotropic hypogonadism have classically been categorized as those without anosmia (termed idiopathic hypogonadotropic hypogonadism, or IHH) and those with IHH and associated anosmia. This latter group is said to have Kallmann syndrome (KS). The prevalence of IHH and KS is estimated to be 1:10,000 to 1:60,000 with a male to female ratio of 5:1. The number of patients who can correctly be termed to have idiopathic disorders is decreasing as investigators identify an increasing number of associated genetic defects. Nevertheless, the terms normosmic IHH and anosmic IHH or KS continue to be useful designations. Furthermore, research in this field is demonstrating that mutations in a single gene may not be reproducibly correlated with normal or abnormal smell, blurring the line between those with IHH and Kallmann syndrome.87
{kind=link}
The genes which cause IHH/Kallmann syndrome can be divided into those which control: (1) the development and migration of GnRH neurons, (2) normal pulsatile GnRH secretion, or (3) GnRH responsiveness at the pituitary (ie., mutations in the GnRHR gene) (Figure 4). A few of the best characterized of these genes are described below. Of note, patients with IHH/KS may also have associated developmental defects, demonstrating the diverse function of the genes which cause these syndromes.
Mutations which correlate with abnormal GnRH migration
As a whole, the mutations in this group result in migratory arrest of the GnRH neuronal precursors before they reach their correct position within the arcuate nucleus. As a result, secreted GnRH does not reach the pituitary and is unable to stimulate gonadotropin secretion. The inheritance pattern of Kallmann syndrome can be X-linked (KAL1), autosomal dominant (FGFR1), or autosomal recessive (PROKR2). There are six known forms of Kallmann syndrome which are distinguished primarily by the genetic mutation associated with them.88, 89
Located on the X chromosome, the KAL1 gene encodes anosmin-1, a secreted extracellular adhesion protein. Anosmin-1 directs migration of the GnRH neurons to the arcuate nucleus and olfactory neurons to the olfactory bulb during fetal development. Therefore, mutations in this protein result in both reproductive and olfactory deficits. Mutations of the KAL1 gene are believed to account for approximately 5–10% of patients with Kallmann syndrome. These patients commonly have associated midline facial defects such as cleft palate and renal agenesis, and also may demonstrate neurologic abnormalities such as synkinesia (mirror movements of the hands), cerebellar dysfunction, or deafness. Olfactory testing can be done easily in the office with strong odorants such as ground coffee. Interestingly, many of these patients are unaware of their deficit.90
Mutations in the fibroblast growth factor receptor-1 (FGFR1) gene have also been associated with normosmic IHH or Kallmann syndrome, as well as associated cleft palate and dental agenesis. FGFR1 mutations are inherited in an autosomal dominant pattern and may count for as many as 10% of IHH cases. Interestingly, mutations in the gene which encodes fibroblast growth factor 8 (FGF8) have recently been shown to cause hypogonadotropic hypogonadism, suggesting that FGF8 is an essential ligand for FGFR1 signaling, at least in terms of the development of a normal GnRH system. Furthermore, FGFR1 expression co-localizes with anosmin, suggesting a functional link between these two proteins.91
The interaction of prokineticin-2 (PROK2) with its receptor (PROKR2) is critical for normal development of the olfactory and GnRH neurons and is estimated to be responsible for 5–10% of all cases of Kallmann syndrome. Genotyping in humans as well as the evaluation of transgenic mouse models suggest that mutations in the PROK2 gene or in the PROKR2 gene are inherited in an autosomal recessive manner. Nevertheless, affected patients have been identified in whom only a single copy of one of these genes is mutated, suggesting that they have mutations in an alternate gene and are, in fact, compound heterozygotes.92, 93
Mutations in the CHD7 and NELF genes are also associated with abnormal GnRH migration. Both CHD7 and NELF are expressed in the hypothalamus as well as the anterior pituitary gland. CHD7 is a chromodomain-helicase-DNA-binding protein. In addition to hypogonadism, mutations in this protein result in CHARGE syndrome, a multisystem disorder which includes Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities and deafness.94 The function of nasal embryonic LHRH factor (NELF) remains unclear, but the presence of two putative zinc fingers suggests that it may act as a transcription factor.95
Genes with critical roles in regulation of GnRH secretion
Inactivating mutations in the KISS1R gene show an autosomal recessive pattern of transmission and represent about 5% of cases of normosmic IHH. The key role of the KiSS-1 receptor in the regulation of the onset of puberty was demonstrated by the development of precocious puberty in a patient with a gain-of-function mutation in this receptor which blunts the rate of receptor desensitization.96
Patients with severe obesity and hypogonadotropic hypogonadism have been found to harbor mutations in the genes which encode leptin or its receptor. These mutations are inherited in an autosomal recessive manner and are uncommon, believed to be responsible for less than 5% of IHH.97
The TAC3 gene encodes neurokinin B (NKB), a member of the substance-P family, and is expressed in the arcuate nucleus. The NKB receptor is expressed on GnRH neurons, at least as studied in rodents. Mutations in both NKB and its receptor have been identified in patients with hypogonadotropic hypogonadism. Interestingly, NKB is co-expressed with kisspeptin in the arcuate nucleus and may, therefore, play a role in the control of GnRH secretion in coordination with the kisspeptin–kisspeptin receptor system.98
Mutations in the GnRH receptor gene
Mutations in the gene which encodes the human GnRH receptor account for 3–6% of sporadic cases of normosmic IHH and up to 40% of familial cases of normosmic IHH.99, 100 Approximately 20 partial or complete loss-of-function mutations in the GnRHR gene have been identified. The phenotype of these patients ranges from complete absence of sexual maturation to delayed puberty. The clinical phenotype correlates with LH pulse profiles and response to exogenous GnRH treatment. The majority of GnRHR mutations are missense and impair GnRH action through abnormal receptor expression, ligand binding, G-protein coupling and/or intracellular trafficking to the cell membrane. Inheritance is autosomal recessive with most patients having compound heterozygous mutations. Recent work in this area has focused on the development of pharmacologic chaperones which can rescue dysfunctional GnRH receptor function through normalization of GnRH receptor folding in the endoplasmic reticulum, thereby restoring ligand binding and intracellular signaling.
CONCLUSIONS
The reproductive system is comprised of a complex network of hormones in which GnRH plays a central role. Over the past four decades, great strides have been made in our understanding of GnRH action in both physiologic and pathologic states. Investigators are beginning to unravel the factors required for GnRH neuronal migration and to understand the mechanisms by which pulsatile GnRH secretion initiates puberty and maintains normal adult reproductive function. In addition, new research is demonstrating significant roles for GnRH and GnRHR outside of the hypothalamic–pituitary axis. This research provides the promise of new clinical applications for GnRH analogues including improved cancer treatment
REFERENCES
Conn PM and Crowley, Jr. WF: Gonadotropin-releasing hormone and its analogs. Annu Rev Med 45:391-405, 1994 |
|
Wetsel WC, Srinivasan S: Pro-GnRH processing. Prog Brain Res 141:221-241, 2002 |
|
Schwanzel-Fukada M and Pfaff DW: Origin of luteinizing hormone-releasing hormone neurons. Nature. 338 6211:161-164 |
|
Wierman ME, Kiseljak-Vassiliades K, Tobet S: Gonadotropin-releasing hormone (GnRH) neuron migration: initiation, maintenance and cessation as critical steps to ensure normal reproductive function. Front Neuroendocrinol 32 (1):43-52, 2011 |
|
Schwanzel-Fukada M and Pfaff DW: Origin of luteinizing hormone-releasing hormone neurons. Nature. 338:161-164, 1989 |
|
Silverman AJ, Jhamandas J, Renaud LP: Localization of luteinizing hormone-releasing hormone (LHRH) neurons that project to the median eminence. J Neurosci 7:2312, 1987 |
|
Knobil E: The discovery of the hypothalamic gonadotropin-releasing hormone pulse generator and of its physiological significance. Endocrinology 131:1005-1006, 1992 |
|
Belchetz PE, Plant TM, Nakai Y, Keogh EJ, Knobil E: Hypophysial responses to continuous and intermittent delivery of hypothalamic gonadotropin-releasing hormone. Science 202:631-633, 1978 |
|
Constantin, S: Physiology of the Gonadotropin-Releaseing Hormone (GnRH Neurone: Studies from Embryonic GnRH Neurones). J Neuroendocrinol 23:542-553, 2011 |
|
Wetsel WC, Valença MM, Merchenthaler I et al: Intrinsic pulsatile secretory activity of immortalized luteinizing hormone-releasing hormone-secreting neurons. Proc Natl Acad Sci 89:4149-4153, 1992 |
|
Waldhauser F, Weibenbacher G, Frisch H, Pollaak A: Pulsatile secretion of gonadotropins in early infancy. Eur J Pediatr 137:71-74, 1981 |
|
Blogowska A, Rzepka-Gorska I, Krzyzanowska-Swiniarska B et al: Leptin, neuropeptide Y, beta-endorphin, gonadotropin, and estradiol levels in girls before menarche. Gynecol Endocrinol 17:7-12, 2003 |
|
McCartney CR: Maturation of Sleep-Wake Gonadotropin-Releasing Hormone Secretion Across Puberty in Girls: Potential Mechanisms and Relevance to the Pathogenesis of Polycystic Ovary Syndrome. J Neuroendocrinol 22:701-709, 2010 |
|
Reame N, Sauder SE, Kelch RP, Marshall JC: Pulsatile gonadotropin secretion during the human menstrual cycle: evidence for altered frequency of gonadotropin-releasing hormone secretion. J Clin Endocrinol Metab 59:328-337, 1984 |
|
Nippoldt TB, Reame NE, Kelch RP, Marshall JC: The roles of estradiol and progesterone in decreasing luteinizing hormone pulse frequency in the luteal phase of the menstrual cycle. J Clin Endocrinol Metab 69:67-76, 1989 |
|
Hall JE, Schoenfeld DA, Martin KA, Crowley WF, Jr.: Hypothalamic gonadotropin-releasing hormone secretion and follicle-stimulating hormone dynamics during the luteal-follicular transition. J Clin Endocrinol Metab 74:600-607, 1992 |
|
Hall JE, Lavoie HB, Marsh EE, Martin KA: Decrease in Gonadotropin-Releasing Hormone (GnRH) Pulse Frequency with Aging in Postmenopausal Women. J Clin Endocrinol Metab 85:1794-1800, 2000 |
|
Johnson J, Skaznik-Wikiel M, Lee HJ, Niikura Y, Tilly JC, Tilly JL: Setting the record straight on data supporting postnatal oogenesis in female mammals. Cell Cycle 4:1471-1477, 2005 |
|
Zou K, Yuan Z, Yang Z et al. Production of offspring from a germline stem cell line derived from neonatal ovaries. Nat Cell Biol 11:631-636, 2009 |
|
Herbison AE, Pape JR: New evidence for estrogen receptors in gonadotropin-releasing hormone neurons. Front Neuroendocrinol 22:292-308, 2001 |
|
Hu L, Gustofson RL, Feng H, Leung PK, Mores N, Krsmanovic LZ, Catt KJ: Converse regulatory functions of estrogen receptor-alpha and -beta subtypes expressed in hypothalamic gonadotropin-releasing hormone neurons. Mol Endocrinol 22:2250-2259, 2008 |
|
Shaw ND, Histed SN, Srouji SS et al: Estrogen negative feedback on gonadotropin secretion: evidence for a direct pituitary effect in women. J Clin Endocrinol Metab 95:1955-1961, 2010 |
|
Marshall JC, Case GD, Valk TW, Corley KP, Sauder SE, Kelch RP. Selective inhibition of follicle-stimulating hormone secretion by estradiol. Mechanism for modulation of gonadotropin responses to low dose pulses of gonadotropin-releasing hormone. J Clin Invest 71:248-257, 1983 |
|
Karsch FJ, Weick RF, Butler WR, Dierschke DJ, Krey LC, Weiss G, Hotchkiss J, Yamaji T, Knobil E: Induced LH surges in the rhesus monkey: strength-duration characteristics of the estrogen stimulus. Endocrinology 92:1740-1747, 1973 |
|
Nakai Y, Plant TM, Hess DL et al: On the sites of the negative and positive feedback actions of estradiol in the control of gonadotropin secretion in the rhesus monkey. Endocrinology 102:1008, 1978 |
|
Knobil E: The neuroendocrine control of the menstrual cycle. Rec Prog Horm Res 36:53, 1980 |
|
Ferin M, Rosenblatt H, Carmel PW et al: Estrogen-induced gonadotropin surges in female rhesus monkeys after pituitary stalk section. Endocrinology 104:50, 1979 |
|
Nippoldt TB, Reame NE, Kelch RP, Marshall JC: The roles of estradiol and progesterone in decreasing luteinizing hormone pulse frequency in the luteal phase of the menstrual cycle. J Clin Endocrinol Metab 69:67-76, 1989 |
|
Soules MR, Steiner RA, Clifton DK, et al: Progesterone modulation of pulsatile luteinzing hormone secretion in normal women. J Clin Endocrionol Metab 58:378-383, 1984 |
|
Howlett TA, Rees LH: Endogenous opioid peptides and hypothalamo-pituitary function. Annu Rev Physiol 48:527-536, 1986 |
|
McShane TM, May T, Miner JL, Keisler DH: Central actions of neuropeptide-Y may provide a neuromodulatory link between nutrition and reproduction. Biol Reprod 46:1151-1157, 1992 |
|
Pielecka-Fortuna J, Chu Z, Moenter SM: Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology 149:1979-1986, 2008 |
|
Roa J, Navarro VM, Tena-Sempere M: Kisspeptins in Reproductive Biology: Consensus Knowledge and Recent Developments. Biol Reprod 85:650-660, 2011 |
|
Tena-Sempere M: Kisspeptin signaling in the brain: Recent developments and future challenges. Molecular and Cellular Endocrinology 314:164-169, 2010 |
|
Teles MG, Bianco SD, Brito SD et al: A GPR54-activating mutation in a patient with central precocious puberty. N Eng J Med 358:709-715, 2008 |
|
Gutierrez-Pascual E, Martinez-Fuentes AJ, Pinilla L et al: Direct pituitary effects of kisspeptin: activation of gonadotrophs and somatotrophs and stimulation of luteinizing hormone and growth hormone secretion. J Neuroendocrinol 19:521-530, 2007 |
|
Luque RM, Cordoba-Chacon J, Gahete MD et al: Kisspeptin regulates gonadotroph and somatotroph function in nonhuman primate pituitary via common and distinct signaling mechanisms. Endocrinology 152:957-966, 2011 |
|
Castellnao JM, Navarro VM, Fernandez-Fernandez R et al: Ontogeny and mechanisms of action for the stimulatory effect of kisspeptin on gonadotropin-releasing hormone system of the rat. Mol Cell Endocrionol 257-258:75-83, 2006 |
|
Tena-Sempere M: Roles of kisspeptins in the control of hypothalamic-gonadotropic function: focus on sexual differentiation and puberty onset. Endocr Dev 17:52-62, 2010 |
|
Han SK, Gottsch ML, Lee KJ et al: Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci. 25:11349-11356, 2005 |
|
Rometo AM, Krajewski SJ, Voytko ML, Rance NE: Hypertrophy and increased kisspeptin gene expression in the hypothalamic infundibular nucleus of postmenopausal women and ovariectomized monkeys. J Clin Endocrinol Metab 92:2744-2750, 2007 |
|
Dumalska L, Wu M, Morozova E et al: Excitatory effects of the puberty-initiating peptide kisspeptin and group I metabotropic glutamate receptor agonists differentiate two distinct subpopulations of gonadotropin-releasing hormone neurons. J Neurosci 28:8003-8013, 2008 |
|
Kauffman AS, Gottsch ML, Roa J et al: Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology 148:1774-1783, 2007 |
|
Quennell JH, Howell CS, Roa J, Augustine RA et al: Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology 152:1541-1550, 2011 |
|
Elias CF: Leptin action in pubertal development: recent advances and unanswered questions. Trends Endocrinol Metab 23:9-15, 2012 |
|
Kim HH: Regulation of Gonadotropin-Releasing Hormone Gene Expression. Semin Reprod Med 25:313-325, 2007 |
|
White et al: Second gene for GnRH in humans. Proc Natl Acad Sci USA 95:305-309, 1998 |
|
Skinner DC, Albertson AJ, Navratil A, Smith A, Mignot M, Talbott H, Scanlan-Blake N: Effects of gonadotrophin-releasing hormone outside the hypothalamic-pituitary-reproductive axis. J Neuroendocrinol 21:282-292, 2009 |
|
Hong IS, Cheung AP, Leung PC: Gonadotropin-releasing hormones I and II induce apoptosis in human granulosa cells. J Clin Endocrinol Metab 93:3179-3185, 2008 |
|
Millar, RP: GnRHs and GnRH receptors. Anim Repro Sci 88:5-28, 2005 |
|
Millar RP: GnRH II and type II GnRH receptors. Trends Endocr Metabol 14:35-43, 2003 |
|
King JA et al: Evolutionary aspects of gonadotropin-releasing hormone and its receptor. Cell mol Neurobiology 15:5-23, 1995 |
|
Cheng CK and Leung PC: Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr Rev 26:283-306, 2005 |
|
Khosravi S, Leung PC: Differential regulation of gonadotropin-releasing hormone (GnRH) I and GnRHII messenger ribonucleic acid by gonadal steroids in human granulose luteal cells. J Clin Endocrionol Metab 88:663-672, 2003 |
|
Norwitz EF, Cardona GR, Jeong KH, Chin WW: Identification and characterization of the gonadotropin-releasing hormone response elements in the mouse gonadotropin-releasing hormone receptor gene. J Biol Chem 274:867-880, 1999 |
|
Yu B, Ruman J, Christman G: The role of peripheral gonadotropin-releasing hormone receptors in female reproduction. Fertility and Sterility 95:465-473, 2011 |
|
Cheng CK and Leung PC: Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr Rev 26:283-306, 2005 |
|
Millar RM, Lowe S, Conklin DA et al: A novel mammalian receptor for the evolutionary conserved type II gonadotropin-releasing hormone. Proc Natl Acad Sci USA 98:9636-9641, 2001 |
|
Morgan K, Conklin D, Pawson AJ et al: A transriptionally active human type II gonadotropin-releasing hormone receptor gene homolog overlaps two genes in the antisense orientation on chromosome 1q.12. Endocrinology 144:423-436, 2003 |
|
Cheung LWT, Wong AST: Gonadotropin-releasing hormone: GnRH receptor signaling in extrapituitary tissues. FEBS Journal 275:5479-5495, 2008 |
|
Hapgood JP, Sadie H, van Biljon W, Ronacher K: Regulation of Expression of Mammalian Gonadotrophin Releasing Hormone Receptor Genes. Journal of Neuroendocrionology 17:619-638, 2005 |
|
Singh P, Krishna A, Sridaran R, Tsutsui K: Immunohistochemical localization of GnRH and RFamide-related peptide-3 in the ovaries of mice during the estrous cycle. J Mol Histol 42:371-81, 2011 |
|
Chakrabarti N, Subbarao T, Sengupta A, Xu F, Stouffer RL, Sridaran R: Expression of mRNA and proteins for GnRH I and II and their receptors in primate corpus luteum during menstrual cycle. Mol Reprod Dev 75:1567-1577, 2008 |
|
Skinner DC, Albertson AJ, Navratil A, Smith A, Mignot M, Talbott H, Scanlan-Blake N: Effects of gonadotrophin-releasing hormone outside the hypothalamic-pituitary-reproductive axis. J Neuroendocrinol 21:282-292, 2009 |
|
Chabre O et al: Coupling of the alpha 2A-adrenergic receptor to multiple G-proteins. A simple approach for estimating receptor-G-protein coupling efficiency in a transient expression system. J Biol Chem 269:5730-5734, 1994 |
|
Vrecl M et al: Internalization kinetics of the gonadotropin-releasing hormone (GnRH) receptor. Pflugers Arch 439 (3 Suppl):R19-R20, 2000 |
|
Choi SG, Jia J, Pfeffer RL, Sealfon SC: G proteins and autocrine signaling differentially regulate gonadotropin subunit expression in the pituitary gonadotrope. J Biol Chem (Epub ahead of print), 2012 |
|
Armstrong SP, Caunt CJ, Fowkes RC, Tsaneva-Atanasova K, McArdle CA: Pulsatile and sustained gonadotropin-releasing hormone (GnRH) receptor signaling: does the Ca2+/NFAT signaling pathway decode GnRH pulse frequency? J Biol Chem 284:35746-35757, 2009 |
|
Imai A, Horibe S, Takagi A, Tamaya T: Gi protein activation of gonadotropin-releasing hormone-mediated protein dephyosphoylation in human endometrial carcinoma. Am J Obstet Gynecol 176:371-376, 1997 |
|
Maiti K, Oh DY, Moon JS, Acharjee S, Li JH, Bai DG, Park HS, Lee K, Lee YC, Jung NC, Kim K, Vaudry H, Kwon HB, Seong JY: Differential effects of gonadotropin-releasing hormone (GnRH)-I and GnRH-II on prostate cancer cell signaling and death. J Clin Endocrinol Metab 90:4287-4298, 2005 |
|
Everest HM, Hislop JN, Harding T et al: Signaling and Antiproliferative Effects Mediated by GnRH Receptors After Expression in Breast Cancer Cells Using Recombinant Adenovirus. Endocrinology 142:4663-4672, 2001 |
|
Kaiser UB, Sabbagh E, Katzenellenbogen RA, Conn PM, Chin WW: A mechanism for the differential regulation of gonadotropin subunit gene expression by gonadotropin-releasing hormone. Proc Natl Acad Sci USA 19;92 26:12280-12284, 1995 |
|
Nathwani PS, Kang SK, Cheng KW, et al: Regulation of gonadotropin-releasing hormone (GnRH) and its receptor gene expression by 17β-estradiol in cultured human granulose-luteal cells. Endocrinology 141:1754-1763, 2000 |
|
An BS, Choi JH, Choi KC, Leung PC: Differential role of progesterone receptor isoforms in the transcriptional regulation of human gonadotropin-releasing hormone I (GnRH I) receptor, GnRH I, and GnRH I. J Clin Endocrinol Metab 90:1106-1113, 2005 |
|
Maya-Nunez G, Conn PM: Transcriptional regulation of the GnRH receptor gene by glucocortocoids. Mol Cell Endocrinol 200:89-98, 2003 |
|
Oakley AE, Breen KM, Clarke IJ, Karsch FJ, Wagenmaker ER, Tilbrook AJ: Cortisol reduces gonadotropin-releasing hormone pulse frequency in follicular phase ewes: influence of ovarian steroids. Endocrinology 150:341-349, 2009 |
|
Gregory SJ, Kaiser UB: Regulation of gonadotropins by inhibin and activin. Semin Reprod Med 22:253-267, 2004 |
|
Winters SJ, Moore, Jr. JP: PACAP, an autocrine/paracrine regulator of gonadotrophs. Biol Reprod 84:844-850, 2011 |
|
Speroff L, Fritz MA: Clinical Gynecologic Endocrinology and Infertility, Seventh Edition Chapter 5, 2005 |
|
Raivio T, Falardeau J, Dwyer A, Quinton R et at: Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 357:863-873, 2007 |
|
Broekmans FJ: GnRH agonists and uterine leiomyomas. Human Reproduction 11:3-25, 1996 |
|
Carr BR, Marshburn PB, Weatherall PT et al: An evaluation of the effect of gonadotropin-releasing hormone analogs and medroxyprogesterone acetate on uterine leiomyomata volume by magnetic resonance imaging: A prospective, randomized, double blind, placebo-controlled, crossover trial. J Clin Endocrinol Metab 76:1217, 1993 |
|
Cann CE, Martin MC, Genant HK et al: Decreased spinal mineral content in amenorrheic women. JAMA 251:626, 1984 |
|
Matta WH, Shaw RW, Hesp R et al: Reversible trabecular bone density loss following induced hypo-oestrogenism with the GnRH analogue buserelin in premenopausal women. Clin Endocrinol (Oxf) 29:45, 1988 |
|
Tan O, Bukulmez O: Biochemistry, molecular biology and cell biology of gonadotropin-releasing hormone antagonists. Curr Opin Obstet Gynecol 23:238-244, 2011 |
|
Del Mastro L, Boni L, Michelotti A, Gamucci T et al: Effect of the goandotropin-releaseing hormone analogue triptorelin on the occurrence of chemotherapy-induced early menopause in premenopausal women with breast cancer: a randomized trial. JAMA 306: 269-276, 2011 |
|
Balasubramanian R, Dwyer A, Seminara SB, Pitteloud N, Kaiser UB, Crowley WF Jr.: Human GnRH deficiency: a unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinology 92:81-99, 2010 |
|
Dode C, Hardelin JP: Clinical genetics of Kallmann syndrome. Ann Endocrinol (Paris) 71:149-157, 2010 |
|
Bianco SD, Kaiser UB: The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol 5:569-576, 2009 |
|
Kaplan JD, Bernstein JA, Kwan A, Hudgins L: Clues to an early diagnosis of Kallmann syndrome. Am J Med Genet A 152:2796-2801, 2010 |
|
Villanueva C, de Roux N: FGFR1 mutations in Kallmann syndrome. Front Horm Res 39:51-61, 2010 |
|
Brioude F, Bouvattier CE, Lombes M: Hypogonadotropic hypogonadism: new aspects in the regulation of hypothalamic-pituitary-gonadal axi. Ann Endocrinol (Paris) 71 (Suppl 1):S33-41, 2010 |
|
Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, Seminara SB, Pitteloud N, Zhou QY, Crowley WF Jr.: The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev 32:225-246, 2011 |
|
Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM: CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 48:334-342, 2011 |
|
Xu N, Bhagavath B, Kim HG, Halvorson L et al: NELF is a nuclear protein involved in hypothalamic GnRH neuronal migration. Mol Cell Endocrionol 319:47-55, 2010 |
|
Hwang JS: The genes associated with gonadotropin-releasing hormone-dependent precocious puberty. Korean J Pediatr 55:6-10, 2012 |
|
Beate K, Joseph N, de Nicolas R, Wolfram K: Genetics of isolated hypogonadotropic hypogonadism: role of GnRH receptor and other genes. Int J Endocrinol, 2012 |
|
Topaloglu AK, Reimann F, Guclu M, et al: TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet 41:354-358, 2009 |
|
Bianco SDC, Kaiser UB: The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Endocrinology 5:569-576, 2009 |
|
Kim HG, Pedersen-White J, Bhagavath B, Layman LC: Genotype and phenotype of patients with gonadotropin-releasing hormone receptor mutations. Front Horm Res 39:94-110, 2010 |