Disorders of the Adrenal Cortex
Authors
INTRODUCTION
Increased or decreased production of steroids (mineralocorticoids, glucocorticoids, or androgens) reflects a disorder of the adrenal cortex. This chapter discusses congenital adrenal hyperplasia (increased androgen production), Cushing's syndrome (increased glucocorticoid production), adrenal insufficiency, and adrenal tumors.
CONGENITAL ADRENAL HYPERPLASIA
Congenital adrenal hyperplasia (CAH) is a group of common inborn errors of metabolism that are transmitted as autosomal-recessive traits.1, 2 These enzymatic deficiencies cause impaired cortisol production as a result of their intermediary role in the steroidogenic pathway that converts cholesterol to cortisol. When cortisol production is decreased, the level of adrenocorticotropic hormone (ACTH) rises because of the absence of negative feedback.3 Because the body attempts to maintain normal cortisol levels, there is overproduction and accumulation of cortisol precursors proximal to the enzymatic block. These precursors are shunted toward the production of C19 androgens (androstenedione and testosterone), leading to the clinical manifestations of virilization and hirsutism.4, 5 The 21-hydroxylase (21-OH) and 11β-hydroxylase enzymes also are used in the mineralocorticoid pathway the end product of which is aldosterone. When the enzyme is defective in this pathway, patients cannot conserve sodium. This condition leads to the salt-wasting form of the disease.
Patients with the classic neonatal forms have sexual ambiguity, progressive virilization, and sometimes salt loss or hypertension. These conditions require life-long medical management6 and often surgical reconstruction of the deformed genitalia.7 The nonclassic (asymptomatic) and late-onset (usually pubertal) forms are more common than the classic presentation. The most common form of classic CAH is 21-hydroxylase deficiency (CAH21), which accounts for 95% of the cases of CAH. The next most common forms are 3β-hydroxysteroid dehydrogenase deficiency (CAH3β-HSD) and 11β-hydroxylase deficiency (CAH11β).8, 9
21-Hydroxylase deficiency
In CAH21, the abnormality is in the 21-OH enzyme (P45021), blocking the conversion of hydroxyprogesterone (17OHP) to 11-deoxycortisol (compound S). Basal circulating levels of both progesterone and 17OHP are elevated in the classic form, but usually only after ACTH stimulation in the cryptic, heterozygote, or attenuated (late-onset) varieties of CAH21.
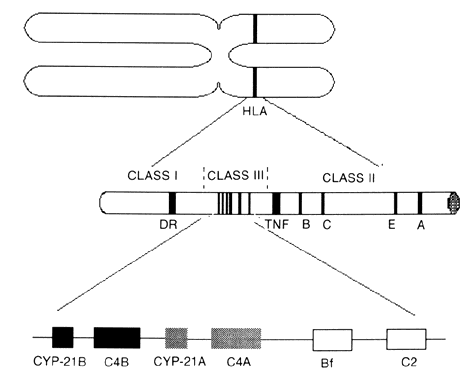
GENETICS
The different types of CAH21 can be attributed to multiple allelic variations encoding the cytochrome P450 enzyme, which performs 21-hydroxylation (CYP-21). Molecular studies showed two copies of the gene on the short arm of chromosome 6p21.3 in the class III region of the human lymphocyte antigens (HLA) complex (Fig. 1). The HLA genetic region encodes genes that have multiple allelic variants that determine histocompatibility. Other genes are located in the class III region, including the 21-hydroxylase genes, two complement factor 4 genes (C4), properidine factor B gene (Bf), and complement factor 2 gene (C2). The two 21-hydroxylase genes (CYP-21A and CYP-21B) alternate with the two genes that encode C4. The class III region of the HLA complex is located between the HLA class II region DR gene and the class I region tumor necrosis factor and HLA-B genes.10, 11, 12, 13 The CYP-21B gene is functionally important, but the CYP-21A gene is a pseudogene; the gene product is enzymatically inactive.13, 14, 15, 16
{kind=link}
Gene deletions and substitutions in the CYP-21B gene affect P45021 function. Because the HLA genes are located in the same portion of chromosome 6 as the CYP-21 genes, certain allelic HLA variants are inherited together (genetically linked) with specific CYP-21 mutations.17, 18 Werkmeister and colleagues15 showed that 25% of patients with classic CAH carried alleles that had CYP-21B gene deletions, despite varied HLA compositions. The remaining 75% had smaller de novo point mutations that caused amino acid substitutions in the CYP-21B gene, thereby rendering the enzyme nonfunctional.
The vast majority of the mutations are caused by gene conversions from CYP-21A to CYP-21B.19, 20 Deletions or further duplications of the C4 and CYP-21 genes can occur as a result of unequal crossover due to misalignment of close spacing of homologous genes during meiosis.15, 21 Triplication of the C4 and CYP-21 genes most frequently is associated with the late-onset form of CAH21.22 The B14 DR1 haplotype that is associated with 78% of cases of late-onset CAH21 features an extra C4B and CYP-21A gene as well as a valine-to-leucine mutation at codon 281 of the CYP-21B gene that normally is present in the CYP-21A gene. This codon in the CYP-21B gene is conserved in several mammalian species.19, 23 The resultant amino acid change promotes a break in an α-helix that is required for optimal enzyme function. Many other mutations transferred from the CYP-21A to the CYP-21B gene have been identified causing the wide variety of phenotypes.19, 20 Most patients are compound heterozygotes having different mutations on each chromosome. The phenotype of the patient is most likely to be consistent with the less severe of the genotypes since the gene product allows for enzyme activity.4, 20
CLINICAL PRESENTATION
Depending on the severity of the P45021 defect, the presentation of CAH can vary from the severe classic virilizing form to the late-onset form, in which hirsutism develops at puberty, to the cryptic (asymptomatic) form, which can be detected only by genetic linkage studies or provocative hormonal testing.4, 24 In the classic form, the female neonate displays profoundly virilized external genitalia, include clitoromegaly and labial fusion due to prenatal elevation of the undulating levels of testosterone (T) and dihydrotestosterone (DHT). However, the ductal structures are female in phenotype, featuring normal cervix, uterus, fallopian tubes, and ovaries. In contrast, boys with this disorder display normal external genitalia at birth. Adolescent males may present with painful testicular rest cell tumors which are composed of adrenal like tissue and are typically bilateral.25 However, both sexes are affected by the high circulating levels of androgens manifested by precocious puberty and early fusion of the epiphyses, resulting in short stature.26, 27, 28
In 75% of the patients with classic CAH21, severe aldosterone deficiency leads to sodium depletion.29 This salt-wasting form of CAH21 can be attributed not only to cortisol deficiency but also to the enzymatic defect at the level of the zona glomerulosa, where the sodium-retaining steroid aldosterone is the major product. In the simple virilizing form, the enzyme functions abnormally only in the zona fasciculata that is responsible for cortisol production.
The patients with the late-onset form of CAH21 have varying degrees of precocious puberty, hirsutism, cystic acne, clitoromegaly, short stature, male pattern baldness, anovulation, oligomenorrhea, and infertility.5, 30, 31 Virilization is rare. The frequency of late-onset CAH21 deficiency in hirsute and PCOS women ranges from 0% to 30%.32, 33, 34 Hirsute patients diagnosed with CAH were more likely to have severe hirsutism, virilization, short stature, family history of hirsutism, earlier onset of symptoms, and regular menses.
INCIDENCE
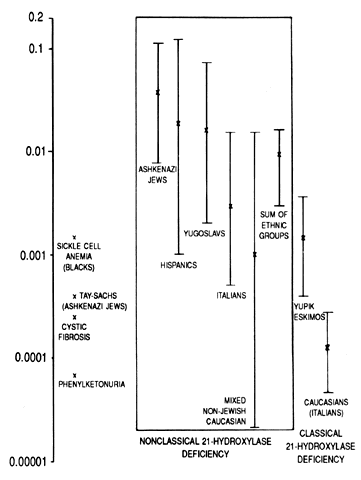
Heel capillary screening of newborns has been performed with a 17OHP assay with a microfilter paper method that was developed by Pang and associates in 1977 and has been used for population screening.35 As calculated by worldwide screening of newborns, there is a 1:60 incidence of heterozygous carriers with a gene frequency of 0.0082. This finding excluded studies in two populations in which the gene frequency was elevated (the Yupik Eskimos of southwest Alaska and the population of La Reunion, France). Infants who had clinical or biochemical evidence of CAH21, such as hyponatremia (serum sodium level less than 130 mEq/L) with associated hyperkalemia (>6.5 mEq/L) or acidosis (HCO3 <17 mEq/L) were classified as having the salt-wasting form. With these criteria, the incidence of the salt-wasting form is three times higher than that of the simple virilizing form. Obviously, neonatal screening programs cannot determine the incidence of late-onset CAH21 because the baseline circulating hormone levels of these patients are normal. By studying the haplotypes of the parents of affected persons and determining whether they are heterozygous or homozygous for 21-OH deficiency by hormonal studies, the incidence of the occurrence of late-onset genes was determined in several populations. Ashkenazi Jews (3.7%), Hispanics (1.9%), Yugoslavs (1.6%), and Italians (0.3%) have a high incidence of attenuated CAH21. In a mixed group of white persons, only 0.1% of affected subjects were homozygous. With sib-pair analysis rather than parental analysis, a similar result was obtained (Fig. 2).4, 36
{kind=link}
HORMONAL DIAGNOSIS
Classic CAH21 is diagnosed by documenting basally elevated circulating levels of 17OHP. Premature, severely ill, and low-birth-weight infants have a higher false-positive rate in the screening programs and cut-off values need to be appropriately adjusted.29, 37, 38 The circulating levels of dehydroepiandrosterone (DHEA), androstenedione, and T39 also are elevated because hormone production is shifted toward androgen biosynthesis. Interestingly, the circulating levels of pregnenolone and 17α-hydroxypregnenolone also can be elevated in classic CAH21; an increase can be explained by either oversaturation of the capacity of 3β-HSD or inhibition of 3β-HSD by the accumulation of its products.40, 41 These precursors are converted to progesterone and 17OHP by the steroidogenic enzyme 3β-HSD.
In late-onset CAH21, basal endocrine determinations may or may not show an increase in the circulating levels of progesterone, 17OHP, androstenedione, T, DHEA, dehydroepiandrosterone sulfate (DHEAS), 11-desoxycortisol, or combinations thereof.40 Significantly, the variable occurrence of basal elevations of these hormones is in large part due to their diurnal (and episodic) fluctuation. Consequently, the timing and frequency of sampling may determine the productivity of basal endocrine testing.
In view of the variable utility of basal endocrine testing, standardized ACTH stimulation tests have been used in recent years.41, 42, 43, 44, 45 These tests usually involve the measurement of steroid precursors proximal to the enzymatic block before and after ACTH stimulation. This approach is based on the premise that subtle enzymatic deficiencies may be brought to light under circumstances that challenge the adrenal steroidogenic machinery. Although in some cases dexamethasone preparation the night before testing has been used, current experience indicates that dexamethasone preparation does not alter corticoid and androgen responses to ACTH.46 The simpler diagnostic test described by Gutai and colleagues43 does not involve dexamethasone preparation. The test assesses the combined rate of increase of progesterone and 17OHP levels over a 30-minute testing period (Fig. 3). A value of greater than 6.5 ng/dl/min is abnormal.
{kind=link}
It is necessary to test female patients during the follicular phase, thereby avoiding the confounding effect of the luteal rise in the circulating levels of progesterone. By measuring 17OHP 60 minutes after administration of 2.5 mg ACTH, New and associates47 developed nomograms to distinguish patients with classic, late-onset, asymptomatic, and heterozygous CAH21 from a control population (Fig. 4). The high incidence of disease in the control population may account for the overlap in values in the nomograms. Elevations in ACTH-stimulated androstenedione level and 17OHP:11-DOC ratio confirm the diagnosis.48 However, if a baseline 17OHP value is greater than 500 ng/dl, late-onset CAH21 can be confirmed without further testing. Some recommend ACTH testing only when a morning 17OHP level is greater than 6 nmol/l.49, 50 Others have shown that basal endocrine determinations cannot identify patients with late-onset CAH21.51
{kind=link}
PRENATAL DIAGNOSIS
Prenatal diagnosis of classic CAH21 allows for early treatment or abortion of an affected fetus and reassurance of the parents of unaffected fetuses. It was first determined in 1975 that 17OHP levels measured in the amniotic fluid could predict classic CAH21 at 24–28 weeks.52 Therefore, second-trimester diagnosis can be made by amniocentesis. Control values differ significantly between laboratories; therefore, each laboratory must standardize its own values. Amniotic fluid levels of androstenedione and 21-deoxycortisol also are elevated and T is in the normal male range in female fetuses with CAH21. Steroid analysis of amniotic fluid cannot be used to diagnose the late-onset form, heterozygotes, or prenatally treated fetuses because amniotic fluid hormone levels are normal in these conditions.53, 54, 55, 56
When the genetic linkage of CAH21 to HLA genes was acknowledged, methods were developed to perform HLA typing on amniotic fluid cells and chorionic villus cells 57 to compare them with an affected sibling HLA type and the parental HLA type.54, 55, 56, 58, 59 HLA typing requires polymorphic differences between the index case and each parent to identify which haplotypes in the parents are responsible for the transmission of an abnormal gene. Sometimes, there is not enough difference in the parent haplotype to determine which haplotype is abnormal; in these cases, HLA typing is nondiagnostic. Errors in predictions for CAH21 can be attributed to recombinations, amniotic fluid contamination with maternal lines, and incorrect HLA typing. Predictions were correct by HLA typing and steroid analysis in 85% of cases.59
After the CYP-21 genes were sequenced,14, 60 a cDNA probe was developed that can identify some deletions in the gene by the absence of a 3.7-kb band that is present in normal persons when Taq1 is used as a restriction endonuclease.61 This method was performed earlier than HLA typing because it did not rely on gene expression, but on the actual DNA.
Rapid molecular genetic screening methods on chorionic villus or amniotic fluid cells are the preferred method of prenatal diagnosis allowing for rapid diagnosis and treatment if needed.37, 62, 63, 64
3β-Hydroxysteroid dehydrogenase deficiency
Although less common than CAH21, CAH3β-HSD has similar presentations. A rare salt-wasting classic form of CAH3β-HSD was described in 1962.65 Infants present with vomiting, feeding problems, dehydration, hyponatremia, and hyperkalemia. In male infants, incomplete masculinization occurs because of inadequate testicular production of testosterone and other androgens. Mild defects such as hypospadias or more significant pseudohermaphroditism with external female genitalia may be present. On the other hand, female infants may have mild virilization due to excess DHEA.66
Hirsutism, acne, and menstrual irregularities occur in women with the late-onset or nonclassic form of CAH3β-HSD caused by the peripheral conversion of the Δ5 steroids to Δ4 steroids in situ at the target organ level appearing like polycystic ovarian syndrome. An allelic variant that causes a late-onset (pubertal, attenuated) form is becoming more commonly recognized in patients who are hirsute, have acne, and have a PCO-like condition.67 Some patients have precocious puberty, menstrual irregularities, or varying degrees of virilism. The estimated incidence of CAH3β-HSD in hirsute women appears to be 12.9%, or approximately one in eight hirsute women.68 Lucky and associates69 studied a group of women with acne and found CAH3β-HSD in 10%. Newer studies incorporating genetic testing did not find abnormalities in patients meeting hormonal criteria for the diagnosis.66, 70 These patients are more likely to have dysregulation of the 17,20 desmolase activity.
The 3β-HSD enzyme converts Δ5 steroids to Δ4 steroids in both the ovary and adrenal gland. Therefore, unlike CAH21, CAH3β-HSD affects both organs. 3β-HSD is not a cytochrome P450 protein and is not HLA linked. Two genes, type 1 and type 2, have been located on chromosome 1p11-13.71, 72 The enzyme functions in the endoplasmic reticulum and mitochondria, and belongs to the aldo-keto reductase group of enzymes as opposed to the P450 enzyme family like the majority of enzymes in the cholesterol steroid pathway.66 Type 1 3β-HSD is expressed in human skin, mammary glands, and adipose tissue, whereas type 2 3β-HSD is expressed in the ovary and adrenal gland.73
Genetic mutations in the 3β-hydroxysteroid dehydrogenase gene have been identified and include early stop codons, frameshifts, and point mutations causing the salt wasting of the disorder due to a nonfunctional enzyme.66, 74, 75 Milder forms are caused by missense mutations that lead to changes in single amino acids which decrease the affinity of the enzyme to its receptor.66, 75 The type 1 3β-HSD gene is preserved in these patients .
Most patients with classic CAH3β-HSD have the salt-wasting form, but a few have normal aldosterone production. There is a 3β-HSD defect in the 17-deoxysteroid pathway in the zona fasciculata in both classic forms, but in the salt-wasting form, the 17α-hydroxysteroid function also is nonfunctional in the zona glomerulosa. Patients with classic CAH3β-HSD have elevated basal and ACTH-stimulated pregnenolone, 17α-hydroxypregnenolone, and DHEA levels. Unexpectedly, Δ4 steroids that are produced past the enzyme block also have slightly elevated levels. However, the ratio of Δ5 to Δ4 steroids is greatly increased. The conversion of DHEA to androstenedione, testosterone, and DHT in the skin and hair follicles was shown previously.76, 77, 78 The elevation of Δ5 to Δ4 steroids is caused by peripheral and liver conversion, which is consistent with the genetic defect in the type 2 mRNA 3β-HSD transcript that is expressed in the ovary and adrenal gland, whereas a normally functioning type 1 mRNA 3β-HSD transcript is expressed in peripheral tissues.73
ACTH stimulation is needed to determine the abnormal hormone function in late-onset CAH3β-HSD. New diagnostic criteria have been proposed since studies have become available which can correlate the genetic defects with the enzyme abnormalities. Lutfallah et al. proposed peak ACTH stimulated levels of 17α-hydroxypregnenolone greater than 290 nmol/l before a diagnosis is made.79 Newer studies have confirmed using these higher levels.80 Using these stricter dianostic critieria, it is rare for the these hormonal abnormalies to be found in hyperandrogenic women.81 Heterozygotes may not have hormonal abnormalities and need to have genetic studies performed to identify unaffected carriers.82
11β-Hydroxylase and related deficiencies
Fewer than 10% of patients with CAH have CAH11β. It has allelic variants that cause classic and late-onset forms. Some patients have hypertension because of elevation of the circulating levels of 11-DOC (precursor at blocked enzyme), which promotes sodium retention, plasma volume expansion, and suppressed plasma renin activity. Other factors may contribute to the hypertension because normalization of the 11-DOC level does not always rectify the hypertension.9
There are two homologous genes on chromosome 8q24.3, the CYP11B1 and the CYP11B2 gene. Therefore, they are not linked to the HLA system, but are located near MYC, NOS, and the thyroglobulin gene.83 These proteins are cytochrome P450s that function in the mitochondria. The CYP11B1 gene product only has 11β-hydroxylase activity, whereas the CYP11B2 gene product performs several enzyme functions: 11β-hydroxylase activity, 18-hydroxylase activity (corticosterone methyl oxidase I [CMO-I] activity), and 18-dehydrogenase activity (corticosterone methyl oxidase II [CMO-II] activity). The isoenzymes are 93% identical.84 The last function is performed only in the zona glomerulosa. CMO-II deficiency is an allelic variant that results from mutations in the CYP-11 structural gene.85
A high rate of CAH11β is found in Moroccan Jews. Many of these families have a single base change in the CYP11B1 that causes a substitution of histidine for arginine, which is conserved in all known eukaryotic P450 proteins.86 Twenty other gene mutations have also been identified in CYP11B1.9 Rarely, mutations in the CYP11B2 gene are associated with isolated aldosterone deficiency patients who present with vomiting and dehydration due to sodium loss and potassium retention. These patients typically are infants or children with normal cortisol and sex steroid levels but elevated renin levels. The highest frequency is found in Iranian Jews.87
The clinical presentation of patients with CAH11β (CYP11B1) is indistinguishable from that of patients with CAH21, with the notable exception of the coexistence of systemic hypertension in some patients. Basal testing typically shows elevated circulating levels of 11-DOC as well as progesterone, 17OHP, and C19 androgens in the classic form.
Diagnosis of late-onset CAH11β is established by an elevated ratio of 18-hydroxycorticosterone to aldosterone in the serum. Maroulis and associates88 evaluated the possible occurrence of CAH11β in a population of hirsute patients and concluded that it is not a common cause of hirsutism. However, several other investigators maintain that appropriate evaluation of this possibility will result in a measurable incidence of CAH11β in hyperandrogenic patient populations.89, 90, 91, 92, 93 Lucky and colleagues69 found evidence of CAH11β in two of 31 patients with acne.
Prenatal diagnosis of classic CAH11β can be made by detection of elevated levels of 11-DOC94 in amniotic fluid or elevated maternal levels of urinary tetrahydrodeoxycortisol (THS) after 8 weeks of gestation. THS is a urinary metabolite of 11-DOC that is not detected in the urine of normal pregnant women but increases progressively in pregnant women with a fetus affected by CAH11β.95
Congenital lipoid adrenal hyperplasia
Inability to perform side chain cleavage of cholesterol is a rare, usually fatal disorder during infancy in which the first step in steroidogenesis is blocked. Profound adrenal insufficiency is present. At autopsy, the adrenal glands are large and filled with lipid which is caused by the accumulation of cholesterol esters.96 Regardless of the genotype, the majority of infants are phenotypic females. A milder form presenting at 2–4 years of age in phenotypically normal males has recently been identified.97 Some female patients have spontaneous puberty since ovarian function is quiescent until puberty and may not be affected by the damage caused by the accumulation of cholesterol esters in well treated patients.98
Initially it was thought to be caused by a mutation in the gene encoding P450SCC (CYP11A1) leading to a deficiency of the 20,22-desmolase enzyme that cleaves choleterol into pregnenolone. A complete lack of enzyme function would lead to lack of progesterone production in the placenta and is likely fatal in utero. Only heterozygotes or compound heterozygotes with partial enzyme function have been identified.99
More often the gene defect is caused by mutations in the steroidogenic acute regulatory protein (StAR) located on chromosome 8p11.2.96, 100 This mitochodrial enzyme is responsible for assisting the transport of cholesterol from the outer to the inner mitochondrial membrane for cholesterol biosynthesis. The protein is found in the ovary, testes, and adrenal gland but is not present in the placenta, therefore placental production of hormones is unaffacted.101
17α-Hydroxylase deficiency
A rare form of congenital adrenal hyperplasia, 17α-hydroxylase deficiency usually is diagnosed around the time of puberty. The involved P450 enzyme that catalyzes 17α-hydroxylase and 17,20-lyase reactions is not necessary for mineralocorticoid synthesis, but is essential for the formation of cortisol, androgens, and estrogens.102
The CYP-17 gene is located on chromosome 10. Four allelic variants that result in nonfunctional P45017 enzyme have been identified. They include a duplication, a substitution, and two deletions.103 At puberty, these patients are sexually immature girls with hypertension and hypokalemia. The high levels of ACTH increase the synthesis of 11-DOC and corticosterone, and produce sodium retention and potassium depletion. Although aldosterone synthesis is unimpaired, the secretion of aldosterone is decreased because the renin–angiotensin system is depressed. Plasma cortisol and gonadotropin levels are elevated. In general, the hormonal picture is the same in male and female patients.
A variant of CAH that has both abnormalities in the activity of 17α-hydroxylase and 21 hydroxylase deficiency has been identified which is caused by mutations in the electron donor enzyme P450 oxidoreductase. In addition to severe genital ambiguity in both sexes, skeletal malformations are also found.8
Treatment of congenital adrenal hyperplasia
Treatment consists of providing replacement levels of glucocorticoids.2, 6 In the classic form, where there is complete enzyme blockage, it is reasoned that providing the equivalent of the daily secretion rate of cortisol by exogenous means will reduce the excessive secretion of ACTH to levels normally observed under physiologic concentrations. As a result, ACTH-driven adrenal androgen production should diminish, and endogenous cortisol secretion should stop. The total daily dose administered should provide the equivalent of the daily cortisol secretion rate to mimic, as much as possible, physiologic cortisol secretion and achieve homeostatic regulation of ACTH release.
The daily cortisol secretion rate in normal adults averages 12.5 ± 2 mg/m2/day or 15–20 mg/day.104 However, if cortisol is given orally, in the form of hydrocortisone tablets, the overall replacement dose may have to be at least doubled because a significant portion of the drug is likely to be inactivated by gastric acidity. Therefore, the daily maintenance dose of oral cortisol is approximately 25 mg/m2/day.105 For a woman of average size (height 5'5” [165 cm], weight 110 lb [50 kg]), the calculated surface area of 1.5 m2 would indicate a daily replacement dose of hydrocortisone of 37.5 mg/day.
If a synthetic glucocorticoid (e.g., dexamethasone, prednisone) is used, several additional factors come into play, complicating the calculation of the optimal replacement dose. Primarily, there is no reliable database with which to compute the daily replacement dose of a synthetic glucocorticoid preparation that would be equivalent to cortisol 37.5 mg/day. Reports of the relative potencies of a variety of glucocorticoid preparations vary widely. For example, dexamethasone has been estimated to have a glucocorticoid effect 25–100 times more potent than that of hydrocortisone. Clearly, such variable estimates complicate the decision about the optimal replacement dose. Although all glucocorticoids can induce pseudotumor cerebri, that tendency appears to be exaggerated with some of the synthetic preparations. Nevertheless, given the cumulative clinical experience, a total daily dose of dexamethasone of 0.5–0.75 mg usually is adequate for the task. Nightly administration for the inhibition of the nocturnal ACTH surge is desirable.106 Likewise, total daily doses of prednisone of 5–7.5 mg probably are equally effective.
Patients must be observed to ensure that the levels of adrenal androgens have been reduced. It is particularly helpful to follow the steroid that is most proximal to the enzymatic block that tends to accumulate in the entity in question (Table 1).107 The regimen given above is not intended for total pituitary adrenal suppression.107, 108, 109, 110 Nevertheless, some patients may be more sensitive than others to the effects of exogenous glucocorticoids, and they may experience undesirable significant suppression of the pituitary–adrenal axis. The ACTH–adrenal axis of these patients may not be able to respond to stress in the form of accident, surgery, or infection, and temporary augmentation of the exogenous glucocorticoid dose may be needed to prevent adrenal crisis. Patients who are taking exogenous glucocorticoids should follow the precautions described for Addison's disease. However, patients treated according to this regimen are not likely to have complete adrenal insufficiency or to require such extreme measures.111, 112 To test for this possibility, a standard ACTH test can be performed.113 If the cortisol response is normal, the patient and physician do not need to be concerned. However, if the ACTH stimulation test result shows complete suppression, the physician should consider lowering the dose. In some patients, complete pituitary adrenal suppression is necessary to achieve control. Under those circumstances, the standard precautions used for patients with Addison's disease should be followed. According to some investigators, suppression of morning basal cortisol levels to less than 2 μg/dl is excessive and requires dose modification.
Table 1. Hormone concentrations measured to diagnose and monitor therapy of the various enzymatic defects that cause congenital adrenal hyperplasia
Enzyme Defect | Elevated Plasma Steroid Level | Elevated Urinary Steroid Level |
21-Hydroxylase deficiency | 17-Hydroxyprogesterone (17-OHP) | Pregnane-3α,17α,20α-triol (pregnanetriol), pregnane-3α,17α,20α-triol-11-one (pregnanetriolone), 17-hydroxypregnanolone |
11-Hydroxylase deficiency | 11-Desoxycortisol (S) | Tetrahydro-11-desoxycortisol (THS) |
3β-Hydroxysteroid dehydrogenase deficiency | Δ5-Pregnene-3β-ol, 20-one (Δ5-pregnenolone), dehydroepiandrosterone (DHEA) | Δ5-Pregnene-3β,17α,20α-triol (Δ5-pregnenetriol) |
(Knorr D, Bidlingmaier F, Kuhle U: Diagnosis and monitoring of therapy of the various enzymatic defects causing congenital adrenal hyperplasia by semiautomatic capillary gas-liquid chromatography. Horm Res 16:201, 1982)
A potent mineralocorticoid, 9α-fluorohydrocortisone or fludrocortisone (Florinef, Squibb Mark, Princeton, NJ; 150 μg/m2/day, 0.2 mg/day), should be started when salt wasting develops in infants with CAH. Salt supplementation of 2.2 mmol/kg/24 hours may be required in the acute phase. Plasma renin levels should be observed to ensure that they are normal. Excessively large amounts of hydrocortisone may be needed if fludrocortisone is insufficient.114 Inadequate replacement can cause failure to thrive.
Growth and devlopment must be monitored closely in children to prevent early epiphyseal closure caused by inadequated treatment and suppressed growth that may be caused by glucocorticoid excess.4 Growth hormone and lupron may be considered as adjunctive treatments to improve final height.115 Obesity may be one of the long term consequences of treatment that needs to be monitored.116
Extensive clinical experience shows that suppression results in the resumption of menstrual cycles in anovulatory patients.6, 105, 117 Although the reason for the improvement in ovulatory status remains uncertain, a reduction of the total effect of androgen, perhaps at the central as well as the intraovarian level, may be responsible. Clearly, androgens may exert a negative feedback effect on gonadotropin release and also may arrest follicular development. If adequately treated, patients with classic CAH will have normal pubertal progression.105 Fertility rates are compromised in patients with classic adrenal hyperplasia potentially due to psychological factors regarding sexuality.118 Reproductive outcomes are improved with patients treated for nonclassical adrenal hyperplasia.119
Although late-onset CAH is thought to be best treated by glucocorticoid replacement, one study suggests that treatment with cyproterone acetate, a peripheral androgen inhibitor, provides greater clinical improvement in hirsutism scores despite continued abnormalities in androgen profiles.120
Prenatal treatment is controversial. David and Forest121 encouraged treatment with dexamethasone of affected female fetuses because they noted less virilization than in their older, untreated affected sisters. The basis of prenatal treatment presupposes that the fetus has an intact pituitary–adrenal axis controlled by ACTH. Dexamethasone is the drug of choice because its transplacental passage is far greater than that of cortisol.122 Since external genitalia development begins at 8 weeks, treatment must be started prior to a diagnosis of CAH being made in the fetus. Treatment should be discontinued if a male or unaffected fetus is identified by chorionic villous sampling or amniocentesis.4, 37 Marked suppression of the adrenal gland was shown after treatment of affected female fetuses with dexamethasone as assessed by the low levels of estriol (fetal androgen metabolite) in the amniotic fluid and maternal urine.123 A dose of 20 μg/kg prepregnancy weight, provided in three equal doses, is recommended (approximately 5 mg/three times a day). Low-dose dexamethasone treatment during pregnancy was not teratogenic.2, 122, 124
CUSHING'S SYNDROME
Etiology
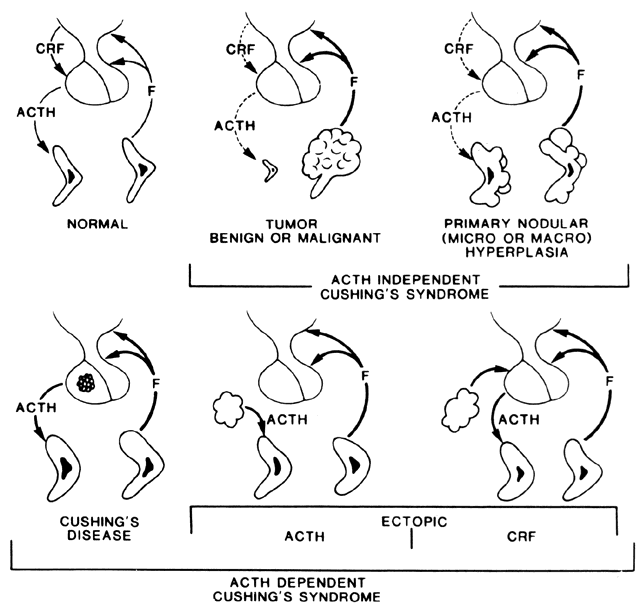
Cushing's syndrome, or hypercortisolism, is an uncommon disorder. Normally, the hypothalamus releases corticotropin-releasing hormone (CRH), which stimulates ACTH secretion by the pituitary gland, which stimulates adrenal cortisol production. Cortisol regulates hypothalamic and pituitary hormonal secretion by negative feedback. Excess cortisol production may occur when there is disruption in the system. It may be an ACTH-dependent process whereby excess ACTH production causes adrenal cortisol hypersecretion. This increase in ACTH secretion may be of pituitary or ectopic origin. A primary adrenal abnormality causes overproduction of cortisol, which is not ACTH dependent and actually causes low levels of circulating CRH and ACTH as a result of feedback inhibition (Fig. 5). Table 2 shows the relative frequency of the various etiologies in the Mayo Clinic experience.125
{kind=link}
The most common cause of endogenous Cushing's syndrome is pituitary hypersecretion of ACTH.126 This variety (also referred to as Cushing's disease) is responsible for approximately 70% (predominantly female) of all cases of Cushing's syndrome. Little is known about the pathogenesis of these adenomas but estrogen may play a role in stimulating the development of these tumors.127
Increased release of CRH by the hypothalamus or increased sensitivity of the pituitary to CRH may be the etiology for some cases of Cushing's disease.128 Rarely, ectopic CRH release129, 130 or ectopic cortisol production131 has been reported. Fifty per cent of ectopic ACTH-secreting tumors are located in the thorax (i.e., oat cell carcinoma, thymoma, bronchial carcinoid). Other possible sources include pancreatic islet cell tumors, medullary thyroid carcinoma, and pheochromocytoma.
Table 2. Frequencies of the various etiologies of Cushing's syndrome
Frequency | Total | ||
Men | Women | ||
Total no. of patients | 108 (34.6%) | 204 (65.4%) | 312 (100%) |
Cushing's disease | 63 (27.8%) | 163 (72.2%) | 226 (72.4%) |
Ectopic ACTH | 19 (55.9%) | 15 (44.1%) | 34 (10.9%) |
Adrenal adenoma | 11 (47.8%) | 12 (52.2%) | 23 (7.4%) |
Adrenal carcinoma | 10 (55.6%) | 8 (44.4%) | 18 (5.8%) |
Nodular adrenal hyperplasia (ACTH independent) | 5 (45.5%) | 6 (54.5%) | 11 (3.5%) |
ACTH, adrenocorticotropic hormone
(Carpenter PC: Diagnostic evaluation of Cushing's syndrome. Endocrinol Metab Clin North Am 17:445, 1988)
Primary bilateral nodular hyperplasia increasingly has been recognized as a cause of Cushing's syndrome. The incidence ranges from 7% to 40% in various series. It is likely due to long term increased stimulation by ACTH causing the nodules. The nodules overproduce cortisol leading to some suppression of the ACTH and therefore produce more cortisol than expected for the given ACTH level.126, 132 There may be several forms of nodular hyperplasia. Some cases are identified in families who have the HLA A1 B8 DR3 haplotype and in whom transmission is thought to be autosomal dominant with variable penetration. In these families, adrenal nodular hyperplasia is associated with cardiac myxoma and spotty pigmentation (lentigo) of the skin. Immunoglobulin G produced by affected siblings can induce cortisol production in guinea pig adrenal glands; therefore, in some patients, nodular adrenal hyperplasia appears to have an autoimmune component.133
Finally, when Cushing's syndrome is associated with true virilization in addition to hirsutism, adrenal carcinoma must be considered a likely cause.126, 134, 135 Adrenal adenomas are less often associated hirsutism but may present with pain and a palpable mass.126 In adults an adrenal etiology occurs in only 10–15% of cases with cancer being very uncommon. However, in children, 65% of cases are of adrenal origin with the majority being due to cancer.136
Clinical presentation
Numerous clinical features may suggest Cushing's syndrome. Progressive central (truncal) obesity, facial rounding (moon facies), and supraclavicular and cervicodorsal fat pads (buffalo hump) representing fat redistribution are the most common manifestations. Changes in protein metabolism cause easy bruising, purpura, and plethoric appearance. Deep red and purple striae, particularly when more than 1 cm wide, suggest hypercortisolism,126 especially when they occur in atypical locations, such as the axillary area, inner thigh, or chest. Progressive hyperpigmentation may occur in patients with Cushing's syndrome associated with high levels of circulating ACTH. It is especially pronounced in patients with ectopic ACTH-producing tumors. ACTH probably plays a primary role in the hyperpigmentation, although over pro-opiomelanocortin (POMC) products may be involved.
Cushing's disease secondary to pituitary hypersecretion of ACTH is most common in women between the ages of 20 and 40 years.135 The clinical symptoms usually are mild, and they progress gradually. In contrast, Cushing's syndrome due to ectopic ACTH production usually has a rapidly progressive course. It occurs most frequently in patients older than 50 years, and classically is accompanied by hypernatremic, hyperchloremic, and hypokalemic alkalosis, and hyperpigmentation. This variety generally is caused by oat cell carcinoma of the lung, pancreatic islet cell carcinoma, or thymoma.137, 138 However, when Cushing's syndrome is caused by a sudden and relatively short-term exposure to hypercortisolism (i.e., carcinoma), patients show little in the way of the Cushing phenotype, which requires chronic glucocorticoid excess.
Hirsutism is a feature of Cushing's syndrome in approximately 60% of patients. The hirsutism typically is moderate, and may be associated with acne and amenorrhea in approximately 40% and 50% of cases, respectively.135, 139 Highly elevated circulating levels of DHEA, DHEAS, testosterone, and androstenedione are suggestive of adrenal carcinoma.140, 141 Cushing's syndrome due to adrenal adenoma usually is not associated with hirsutism, and these patients may have low-normal DHEAS values. Indeed, cortisol-producing adrenal adenomas tend to suppress the pituitary release of ACTH, thereby causing atrophy of the contralateral adrenal gland.
Excess levels of cortisol increase the rate of catabolism, resulting in generalized muscle wasting and weakness that typically is most prominent in the proximal limb muscles. In patients with excess levels of mineralocorticoid, hypokalemia may contribute to the weakness.142 If hypercortisolism is present in childhood, the catabolic effects result in short adult stature. Longitudinal growth of bone commonly is retarded by the excess circulating cortisol, epiphyseal maturation may be accelerated by increased concentrations of circulating adrenal androgens, and abnormal hypothalamic–pituitary function may inhibit the release of growth hormone. Abnormal glucose tolerance test results are present in most patients. The fasting serum glucose level usually is normal, but there is a delayed return to normal after glucose loading. Fractures that occur after minimal trauma and back pain with unsuspected spinal compression fractures are not uncommon143 due to the decrease in protein matrix synthesis by osteoblasts and increased bone resorption. Osteoporosis may be evident radiologically.
Hypertension is a frequent finding in noniatrogenic Cushing's syndrome.127 Although the hypertension rarely is severe, affected persons occasionally have left ventricular hypertrophy and changes in the electrocardiogram result. The mechanisms involved include the salt-retaining action of high levels of circulating glucocorticoids, enhanced vascular reactivity to vasoactive substances such as norepinephrine and angiotensin II, and activation of the renin–angiotensin system.144 Chronic disease also can be associated with an increased risk of vascular occlusion and myocardial infarction. Dependent edema occurs in some patients; it appears to be due to a combination of the salt-retaining action of steroids and a decrease in tissue support.
Psychiatric disturbances are common with more than 70% of the patients presenting with symptoms.127, 145 Especially frequent are irritability, mood disturbances, impaired memory and concentration, insomnia, and a decrease in libido. Thought disorders, including paranoid and confusional states, occur less frequently. These more severe impairments are associated with particularly high concentrations of ACTH and cortisol. Other POMC products may contribute directly to the psychiatric manifestations. In general, the psychiatric disturbances improve after medical or surgical therapy for Cushing's syndrome, although they may persist to a lesser degree for years.
Hematologic manifestations include mild erythrocytosis, granulocytosis, lymphopenia, and eosinopenia. The inhibitory effects of cortisol on leukocyte function and antibody formation predispose patients with Cushing's syndrome to a variety of infections. Life-threatening opportunistic infections are especially likely to occur in patients with high cortisol levels. The most common infections are cryptococcosis, aspergillosis, nocardiosis, and pneumocystosis.146 Historically, infection was the leading cause of death in patients with Cushing's syndrome.135
Diagnosis
The best tests to use, in which patients, how frequently, and in which order have been difficult to determine due to the difficulty in ruling out multiple pseudo-Cushing's states such as depression, alcoholism, obesity, and poorly controlled diabetes.147, 148, 149 After hypercortisolism is established, the specific etiology should be identified (Table 3). Twenty-four-hour urinary excretion of free cortisol (UFC) is increased in patients with Cushing's syndrome and is considered the gold standard for the diagnosis.150 The normal value is less than 80 μg/day. A 24-hour urine collection for 17-hydroxycorticosteroid evaluation also may be performed; the upper limit of normal for this finding is 8 mg/24 hours. Increased salivary cortisol concentrations and midnight plasma cortisol levels have also been used.148 Screening also can be accomplished by the overnight dexamethasone suppression test, which requires a 1-mg oral dose of dexamethasone at 11.00pm followed by determination of plasma cortisol level at 8.00am. A plasma cortisol level of less than 6 μg/dl is considered normal, and a normal value provides good evidence against Cushing's syndrome.151, 152 In contrast, cortisol levels of greater than 10 μg/dl strongly suggest Cushing's syndrome.
Table 3. Summary of the diagnostic evaluation for the various etiologies of Cushing's syndrome
Serum ACTH Level | CRH Test | High-Dose Dexamethasone Test | CT/MRI | Petrosal Sinus Sampling | Iodocholesterol Scan | ||
Sella | Adrenal Glands | ||||||
Pituitary Cushing's | Normal to mildly elevated | + | >50% suppression | ± | Mild bilateral enlargement or normal | + | + |
Ectopic ACTH | Normal to markedly elevated | − | No suppression | − | Enlarged bilaterally or normal | − | + |
Adrenal neoplasms | Undetectable | − | No suppression | − | Unilateral adrenal mass | NA | Adenomas +, carcinomas − |
Micronodular disease | Undetectable | − | No suppression | − | Minimal diffuse enlargement (“knobby”) or normal | − | ± |
ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone;
CT, computerized tomography; MRI, magnetic resonance imaging;
NA, not applicable; +, positive; -, negative; ±, positive or negative
(Perry RR, Nieman LK, Cutler GB et al: Primary adrenal causes of Cushing's syndrome. Ann Surg 210:59, 1989)
A number of pitfalls must be avoided when the overnight dexamethasone test is performed. Patients with depression may not suppress the cortisol level after dexamethasone suppression, and thus may yield a false-positive result.153 Phenytoin (Dilantin, Parke-Davis, Morris Plains, NJ,USA) therapy induces hepatic enzymes, which can lead to a lack of dexamethasone suppression.154 Significantly, estrogen therapy may increase cortisol binding and hence elevate total plasma cortisol values, thereby falsely suggesting a lack of dexamethasone suppression. False-positive findings also occur in 13% of obese patients and 23% of chronically ill patients.
Patients who have morning cortisol values of 6–10 μg/dl after the 1-mg dexamethasone suppression test should have further testing to prove Cushing's syndrome. In patients with Cushing's syndrome, measurement of the serum cortisol or plasma ACTH level at various times during the day will show loss of circadian rhythm. An average evening plasma cortisol value of 10 μg/dl or greater is diagnostic of Cushing's syndrome. To further confirm the diagnosis, the low-dose dexamethasone suppression test generally is performed.155, 156 The patient is given 0.5 mg of dexamethasone orally every 6 hours for 2 days. Suppression of urinary 17-hydroxycorticosteroid excretion to less than 3 mg/24 hours (or to 50% of the baseline value), of the plasma cortisol value to less than 4 μg/dl, or of urinary free cortisol excretion to less than 25 μg/24 hours excludes Cushing's syndrome.126 In contrast, failure to suppress establishes the diagnosis of Cushing's syndrome and necessitates a high-dose dexamethasone suppression test.
The objective of the high-dose dexamethasone suppression test developed by Liddle155 is to differentiate between the ACTH-dependent and ACTH-independent causes of Cushing's syndrome.156 The patient is given 2 mg of dexamethasone orally every 6 hours for 2 days. Suppression of urinary 17-hydroxycorticosteroid excretion to less than 3 mg/24 hours or to less than 50% of the baseline value suggests the diagnosis of pituitary hypersecretion of ACTH. In contrast, failure to suppress may be considered diagnostic of hypercortisolism due to adrenal adenoma, adrenal carcinoma, or ectopic ACTH release.157
Variations of the Liddle high-dose dexamethasone test have been developed to simplify the test and to shorten or eliminate the need for a hospital stay. An overnight 8-mg dexamethasone test158 and a continuous 7-hour intravenous dexamethasone test at 1 μg/hour159, 160 show similar specificity, sensitivity, and diagnostic accuracy when compared with the prolonged Liddle test.
Other tests have been developed to assist in confirming the etiology of Cushing's syndrome. An CRH stimulation test is an expensive but accurate alternative to the dexamethasone suppression test.161 A dose of 1 μg/kg, or 100 μg, CRH is given. Blood samples for ACTH and cortisol are obtained every 15 minutes for 2–3 hours. A 50% increase in plasma ACTH level and a 20% increase in serum cortisol level is considered a positive response, and is diagnostic of pituitary Cushing's syndrome.162, 163 A negative response is obtained in adrenal and most ectopic causes of hypercortisolism. The diagnostic accuracy of CRH testing combined with high-dose dexamethasone was 98% compared with 85–90% with either test alone.164 In ectopic ACTH-producing tumors, no increase in ACTH or cortisol level would be expected with CRH stimulation. However, some lung tumors have shown significant increases in plasma ACTH level after CRH stimulation.165 One explanation is that the tumor is producing CRH instead of or in addition to ACTH.
Plasma ACTH levels are useful in the differential diagnosis of Cushing's syndrome. Patients with Cushing's syndrome secondary to an adrenal tumor usually have low ACTH levels (less than 20 pg/ml).156 Patients with Cushing's disease usually have moderately elevated or high-normal values, whereas patients with ectopic ACTH-producing tumors often have markedly elevated values. However, there is considerable overlap between these two groups.
Imaging techniques
Improved imaging techniques have greatly enhanced the diagnosis of Cushing's syndrome. If a pituitary origin has been established by CRH stimulation and high-dose dexamethasone testing, magnetic resonance imaging (MRI) with gadolinium contrast should be obtained to localize the pituitary adenoma. MRI is more sensitive, specific, and accurate than computerized tomography (CT).162
Some pituitary adenomas are too small to be visualized, even by MRI. Bilateral simultaneous venous sampling of the inferior petrosal sinuses was developed to localize the adenoma with selective catheterization. The inferior petrosal sinus receives the venous blood from the ipsilateral pituitary gland. ACTH levels are higher in the ipsilateral sinus of the microadenoma than in the contralateral side or the periphery. A gradient above 1.4–2.0 is considered significant.166, 167, 168 The gradient can be magnified by CRH stimulation. β-Lipoprotein (β-LPH), a POMC-derived peptide, is secreted with ACTH, and may be assayed to confirm the ACTH results.169 The test is technically difficult, and should be performed only by an experienced neuroradiologist.
An adrenal CT scan is highly effective in localizing an adrenal adenoma or carcinoma. MRI has not been found to contribute accuracy.170 CT evidence of malignant invasion of other structures, tumor size of more than 5 cm, tumor necrosis, and calcification help to distinguish an adrenal carcinoma from adenoma. A normal-sized adrenal gland often is observed in cases of Cushing's disease. Overall increased size of the adrenal gland may suggest ectopic ACTH release, whereas multiple nodules on both sides can be seen in patients with nodular adrenal hyperplasia.171
In addition, CT or MRI evaluation of the lung, mediastinum, and pancreas may assist in the search for an ectopic ACTH source.126, 147
Adrenal nodules are identified as incidental findings in 0.6–9% of all abdominal CT scans. Six of 122 (5%) asymptomatic patients with adrenal adenoma had elevated levels of urinary free cortisol, elevated cortisol levels in overnight dexamethasone suppression tests, loss of diurnal rhythm of cortisol secretion, or low levels of ACTH.172
Adrenal scintigraphy is a noninvasive radiographic technique that identifies functional and structural abnormalities of the adrenal gland. 6-β Radioactive iodine 19-methylnorcholesterol (NP-59) is a radioactive cholesterol precursor of adrenal hormones. Normal adrenal glands show minimal uptake of NP-59 after 5–7 days. Increased NP-59 uptake occurs with bilateral adrenal hyperplasia caused by ACTH-producing pituitary adenomas. An adrenal adenoma will show increased uptake within the adenoma and decreased uptake in the contralateral adrenal gland.173, 174, 175 There is bilateral nonvisualization of the adrenal glands in adrenal carcinoma. Nodular hyperplasia typically shows asymmetric bilateral accumulation of NP-59.176, 177 Since many ectopic ACTH producing tumors have been found to have somatostatin receptors, 111In-labeled octreotide, has been used to detect ectopic ACTH producing tumors.178, 179
Summary
In summary (see Table 3), initial diagnosis is made by determination of elevated urinary free cortisol excretion and low-dose dexamethasone test results. A high-dose dexamethasone test (Liddle's traditional test or a modification) is performed in conjunction with the CRH stimulation test and ACTH level evaluation to determine whether the source of hypercortisolism is pituitary, adrenal, or ectopic. Once the source has been determined, appropriate radiologic tests are performed. If a pituitary source is suspected, an MRI is performed. If no lesion is identified, then inferior petrosal venous sampling is advised. If an adrenal source is suspected, an abdominal CT scan will identify most adrenal lesions. Adrenal scintigraphy can be helpful in determining the functional status of the adrenal glands. An ectopic source should be searched for with the use of MRI or CT scanning of the chest and abdomen.
Treatment
Treatment depends on the etiology of Cushing's syndrome. The treatment of choice for a pituitary adenoma is transsphenoidal surgical removal.180 An immediate cure rate of approximately 90% can be expected when an adenoma is identified on a pathologic specimen. Preoperatively, hypertension and hyperglycemia should be controlled medically, and glucocorticoid coverage should be provided intraoperatively.181 Macroadenoma and extracellular extension are poor prognostic signs. If an adenoma cannot be visualized by MRI, lateralized by inferior petrosal sampling, or identified during surgery, then a total hypophysectomy is suggested for patients who have completed childbearing. If inferior petrosal sinus sampling can identify a gradient between pituitary lobes, then a hemiresection of the hypophysis can be performed. If an adenoma cannot be identified during surgical exploration (in patients who desire future childbearing), then pituitary radiation, adrenalectomy, or medical therapy is advised to preserve gonadotropic function. Complications from surgery include cerebrospinal fluid leaks (1.9%), diabetes insipidus (2.8%), sinusitis (1.9%), and postoperative visual field defects (1.4%). Recurrences of disease can develop up to 8 years after surgery.182, 183
Pituitary radiation can be used to treat recurrences and as an alternative to surgery. Because remission occurs after a median of 4 years, medical therapy is required during the transition period. ACTH production declines after therapy, so hypopituitarism can develop with time.184 With higher dosages, radiation necrosis of the brain can occur years after treatment. It is manifested by localized neurologic deficits.151, 181 Newer techniques such as linear accelerator radiosurgery or gamma knife which more accurately focus the radiation beam on the tumor may have fewer complications and offer better long term results.180, 185
Adrenal surgery can be performed for primary adrenal causes of Cushing's syndrome or for other forms of Cushing's syndrome when other methods fail. Before success was achieved with transsphenoidal pituitary surgery, adrenalectomy was performed, even for patients with Cushing's disease. However, because of the clinical progression of pituitary adenoma, Nelson's syndrome developed after adrenalectomy in approximately 30% of patients with Cushing's disease. Nelson's syndrome is caused by the continuous secretion of ACTH in large amounts without the benefit of negative feedback from cortisol secretion. Patients have hyperpigmentation, visual field defects, headaches, and oculomotor paralysis. Local invasion of the cavernous sinus may occur, and distant metastases have been documented.180, 181
If an adrenal adenoma is located by CT, unilateral adrenalectomy by laparoscopy is advised and there is an almost 100% cure rate.186 Bilateral adrenalectomy is advised for patients with nodular adrenal hyperplasia. If a bilateral adrenalectomy is performed, then mineralocorticoid replacement (Florinef 50–100 mg/day) as well as glucocorticoid replacement is required. For patients with adrenal adenomas, the contralateral adrenal gland is depressed by the negative feedback caused by the adenoma-derived hypersecretion of cortisol. These patients require glucocorticoid replacement until the remaining adrenal gland can maintain adequate steroid production.
Surgical resection of adrenal carcinomas and ectopic ACTH-secreting tumors should be performed if possible. Palliation with medical therapy should be attempted as needed.187, 188
Medical therapy designed to suppress cortisol secretion can be helpful while these patients await surgery, while radiation treatment becomes effective, and for the palliation of carcinomas that produce ectopic ACTH or CRH. Pharmaceutical agents are aimed at decreasing the secretion of ACTH and cortisol. Cyproheptidine has antiserotonin effects and sodium valproate has anti-γ-aminobutyric acid effects that decrease ACTH release.189 SMS 201-995 (octreotide) and SOM230, long-acting analogues of somatostatin, have shown promise in patients with ACTH-dependent Cushing's syndrome.190, 191
Aminoglutethimide, metyrapone, mitotane, and ketoconazole act at the level of various steroidogenic enzymes in the pathway from cholesterol to cortisol production.192, 193, 194 Aminoglutethimide inhibits the action of 20,22-desmolase, which converts cholesterol to pregnenolone. It is only transiently effective in ACTH-dependent Cushing's syndrome because ACTH can override its effect. Side effects include nausea, vomiting, sedation, blurred vision, lethargy, and hypothyroidism. Mitotane inhibits 11β-hydroxylase and 20,22-desmolase in adrenocortical cell mitochondria, causing cellular destruction. Side effects include nausea, anorexia, diarrhea, hypercholesterolemia, and hepatotoxicity. Metyrapone is an 11β-hydroxylase inhibitor that decreases cortisol secretion while increasing 11-DOC and DHEAS secretion. 11-DOC can cause hypertension and hypokalemic alkalosis. The elevation in DHEAS can worsen hirsutism. However, metyrapone has been used as an adjunct to therapy in combination with aminoglutethimide195 or sodium valproate.196
Ketoconazole has been used extensively in the treatment of Cushing's syndrome. Initially, it was developed as an antimycotic drug because it blocks 14-demethylation of lanosterol in fungi. Ketoconazole inhibits cytochrome P450 mono-oxygenases that are involved in steroid biosynthesis.197, 198 Most studies showed no direct effect of ketoconazole on pituitary ACTH release,199, 200 although one in vitro study argued that there may be such an effect.201 Ketoconazole also acts as a glucocorticoid antagonist by competing with dexamethasone at receptor sites.202 Cushing's syndrome is treated with doses of 400–1000 mg/day ketoconazole. Circulating DHEA, DHEAS, androstenedione, cortisol, and testosterone levels decrease with ketoconazole administration, and hirsutism regresses.203, 204 17OHP and progesterone levels increase due to a greater enzyme dysfunction of 17α-hydroxylase.205 Several studies of the use of ketoconazole in Cushing's syndrome showed decreasing cortisol levels.205, 206, 207, 208 This drug even has been used successfully during pregnancy.209 Therefore, ketoconazole is considered a good adjunctive therapy to surgical treatment.
When using ketoconazole, caution must be exercised to avoid hepatic toxicity and hypoadrenalism. Idiosyncratic hepatotoxicity is estimated to occur in 1:15,000 patients. Most often, the elevation in liver function tests (LFT) values is mild and reversible. However, several fatalities have been reported in patients who continued to receive ketoconazole despite abnormal LFT findings.210, 211 McCance and colleagues207 found abnormal LFT results in three of six patients with Cushing's syndrome who received 800 mg/day ketoconazole. The LFT results returned to normal by 15 days after the discontinuation of ketoconazole.
In summary, the primary treatment of Cushing's syndrome should be surgical to remove the source of hormone production, whether it is pituitary, adrenal, or ectopic. Radiation can be used to treat recurrent pituitary abnormalities. Medical treatment is reserved for preoperative treatment, surgical failures, and palliation of incurable disease.
ADRENOCORTICAL INSUFFICIENCY
Etiology
Adrenocortical insufficiency may reflect a primary defect in hormone secretion by the adrenal cortex (Addison's disease) or a secondary pituitary deficiency in ACTH secretion or inhibition of cortisol production by drugs. Regardless of its cause, Addison's disease is associated with weakness, fatigue, and gastrointestinal distress. The incidence of Addison's disease is low; an incidence of 4–6 per million has been reported. Due to the increasing use of steroids, which suppress pituitary ACTH secretion, secondary adrenocortical insufficiency is more common with an incidence of approximately 200 per million. Before the use of antibiotics, tuberculosis was the most common cause of Addison's disease.212, 213 However, the most common etiology now is autoimmune (also referred to as idiopathic) disease. Other granulomatous diseases, such as histoplasmosis, coccidioidomycosis, and cryptococcosis, occasionally are the cause. Twenty per cent of patients with AIDS develop adrenal insufficiency because they may have these diseases as well as CMV, Mycobacterium avium-intracellulare, and Kaposi's sarcoma, all of which affect the adrenal glands. Rarely, bilateral hemorrhage, metastasis, carcinoma, amyloidosis, or sarcoidosis may be the etiology of primary adrenal insufficiency.214, 215 More than 90% of the adrenal glands must be affected before signs of hormonal deficiency become apparent.216 If an enlarged adrenal gland is noted on CT scan in patients with cancer, 20% have or will have adrenal insufficiency.217
Addison's disease can manifest itself at any age. There has been a shift from a slight male predominance to a slight female predominance as the percentage of cases due to autoimmune disease has increased.
Histopathologic examination of the adrenal gland of a patient with idiopathic Addison's disease shows diffuse lymphocytic inflammation in the cortex, with selective destruction of the steroid-producing epithelial cells and sparing of the medulla. Most of these patients have circulating antibodies directed against autoantigens that include constituents of the cytoplasm of adrenocortical cells and also may be expressed on cell surfaces.218, 219 Additionally, evidence has been provided for cell-mediated immune reactions to adrenal antigens. In addition, 60–95% of patients with autoimmune adrenalitis have adrenocytoplasmic microsomal antibodies. Many patients have antibodies to other tissues (i.e., thyroglobulin antibodies, 70%; antiparietal cell or anti-intrinsic factor, 40%), islet cell antibodies (25%), and Sertoli cell antibodies (13%). Evidence of adrenal surface-reactive autoantibodies suggests that complement-mediated humeral cytotoxicity and antibody-dependent cellular cytotoxicity by killer cells may cause the cell damage that is responsible for autoimmune Addison's disease.220
Addison's disease often is found in association with multiple autoimmune disorders. These can be classified as type 1 or type 2 polyglandular autoimmune syndromes (PGA). Type 1 PGA (APECED - autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy) requires at least two of the triad of Addison's disease, hypoparathyroidism, chronic mucocutaneous candidiasis, and usually presents in childhood. Type 1 PGA syndrome displays autosomal-recessive inheritance which is caused by a mutation in the autoimmune regulator gene (AIRE) located on chromosome 21q22.3.221 Type 2 PGA which is more common and is characterized by patients who have Addison's disease with autoimmune thyroiditis or insulin-dependent diabetes mellitus (Schmidt's syndrome) but do not have hypoparathyroidism or candidiasis.222, 223 It has a peak onset in the during the third decade of life. These PGA syndromes and associated autoimmune disorders are compared in Table 4.224 Gonadal failure is present in 5–50% of patients with type 2 PGA syndrome. Ovarian failure is more common than testicular failure. In some cases, gonadal failure is the first manifestation of the syndrome.225, 226 A gene mutation on chromosome 6 and the HLA haplotypes B8 and DR3 have been associated with type 2 PGA syndrome which displays autosomal dominant inheritance. Type 3 PGA is associated with thyroid abnormalities but no adrenal insufficiency and type 4 PGA is associated with adrenal insufficiency and other autoimmune disorders but without thyroid abnormalities.227
Table 4. Polyglandular autoimmune syndromes
Characteristic | Type I | Type II |
Age at onset | Childhood | Adult |
HLA association | None | B8-Dw3-DR3 |
Disease components (incidence) | Addison's disease (67%) | Addison's disease (100%) |
Autoimmune thyroid disease (10–11%) | Autoimmune thyroid disease (69%) | |
Pernicious anemia (13–15%) | Pernicious anemia (<1%) | |
Diabetes mellitus (2–4%) | Diabetes mellitus (52%) | |
Gonadal failure (12–17%) | Gonadal failure (3.5–3.6%) | |
Vitiligo (8–9%) | Vitiligo (4.5%) | |
Hypoparathyroidism (82%) | Celiac disease (ND) | |
Chronic mucocutaneous candidiasis (73–78%) | Myasthenia gravis (ND) | |
Chronic active hepatitis (11–13%) | ||
Alopecia (26–32%) | ||
Malabsorption syndromes (22–24%) |
(Trence DL, Morley JE, Handwerger BS: Polyglandular autoimmune syndromes. Am J Med 77:107, 1984) ND, not determined; HLA, human lymphocyte antigen
Adrenoleukodystrophy is an X linked disorder which presents with neruological abnormalities due to demyelinization and adrenal insufficiency. It is caused by a mutation and the ABCD1 gene located at Xq28 which encodes a peroxisomal membrane protein with variable phenotypes. This mutation causes very long chain fatty acids which can be measured in plasma and chorionic villi.228, 229 Half of the patients present in early childhood and have rapid progression of the disease. A third present later and have a slower progression which only effects the spinal cord and peripheral nerves. A few only develop the adrenal insufficiency.230, 231
Agenesis of the adrenal can occur due to genetic mutations in genes required for the development of the adrenal gland but accounts for only 1% of patients.227 These include mutations in the steroidogenic acute regulatory protein (StAR), 21, 11b and 17 hydroylase, hydroxysteroid dehydrogenase 2, ACTH receptor (MC2R), DHCR7, NROB1, and NR5A1.213
Adrenal hemorrhage is an uncommon etiology of adrenal insufficiency. In children, it is associated with severe meningococcal or Pseudomonas septicemia. Anticoagulation has led to spontaneous adrenal hemorrhage that caused adrenal crisis in more than 50 reported cases. Both warfarin and heparin have been implicated. Interestingly, more than half of these patients have coagulation factor values in the normal range.
Other drugs that affect adrenal hormone production and use have caused adrenal failure. Drugs that inhibit steroid biosynthesis, such as aminoglutethimide, op'DDD, and ketoconazole, have been reported to cause adrenal insufficiency. In patients treated for Addison's disease with steroid replacement, hepatic oxygenase inducers, such as barbiturates, phenytoin, and rifampin, as well as end-organ inhibitors, such as spironolactone, have required an increase in patient dose due to increased metabolism or decreased effectiveness of the steroids.
Secondary adrenal insufficiency also can occur when long-term glucocorticoid therapy is used for replacement doses or pharmacologic treatment of other chronic diseases. Glucocorticoid treatment that suppresses ACTH production causes adrenal atrophy. Therefore, if the exogenous steroid is withdrawn, or stress causes larger dose requirements, the adrenal glands cannot respond, causing secondary adrenal insufficiency.
The pituitary is the primary cause of some cases of secondary adrenal insufficiency. Patients with panhypopituitarism are deficient in ACTH; therefore, glucocorticoid must be replaced.
Clinical presentation
In 1855, Addison's disease was described in 11 patients as a “general languor and debility, remarkable feebleness of the heart's action, irritability of the stomach and a peculiar change of the color of the skin.” This clinical picture was formulated before knowledge of glucocorticoid and mineralocorticoid secretion by the adrenal glands. Table 5 lists the frequency of the common signs and symptoms.232 The onset of symptoms usually is insidious in primary deficiency. Nausea, vomiting, and anorexia are the most common gastrointestinal symptoms, but abdominal pain may be so severe that an acute abdominal disorder is suspected. Occasionally, diarrhea is present.233
Table 5. The major manifestations of Addisons's disease in a study of 108 patients
Findings | Percentage of Patients |
Weakness and fatigue | 100 |
Weight loss | 100 |
Hyperpigmentation | 92 |
Hypotension | 88 |
Hyponatremia | 88 |
Hyperkalemia | 64 |
Gastrointestinal symptoms | 56 |
Postural dizziness | 12 |
Adrenal calcification | 9 |
Hypercalcemia | 6 |
Muscle and joint pain | 6 |
Vitiligo | 4 |
(Nerup J: Addison's disease: Clinical studies. A report of 108 cases. Acta Endocrinol 76:127, 1974)
Hyperpigmentation of the skin and mucous membranes aids in the diagnosis of primary adrenal insufficiency. It is most common on the back of and creases of the hands, elbows, and knees; on the dental gingival margin; on the lips; and on the buccal and vaginal mucosa. These excess melanin deposits range from a slight prolonged tan or an increase in black freckles to an intense generalized pigmentation. Areas exposed to the sun and old scars are affected to a greater degree. Pigmentation is present when an elevated level of ACTH occurs (in primary, but not in secondary disease), and probably is due to β-lipoprotein, which is released in equimolar amounts to ACTH during processing of the POMC molecule. Other POMC products, such as the melanocyte-stimulating hormones, may play a role in hyperpigmentation. Vitiligo caused by autoimmune destruction of melanocytes may be seen in association with hyperpigmentation.
The fatigue that is associated with Addison's disease worsens with activity and improves with rest. Postural hypotension and syncope may occur. Patients usually have normal blood pressure, but the systolic blood pressure rarely exceeds 110 mmHg. Psychological abnormalities also are reported, and probably are due to elevations in POMC products. Decreased axillary and pubic hair may be evident in women with Addison's disease due to decreased adrenal androgen production. This decrease in body hair is not noticeable in men because the testes continue to produce androgens. An uncommon but interesting finding is calcification of cartilage that change the ears to a stone-like consistency. This condition also occurs with acromegaly, sarcoidosis, and ochronosis.
Suspicion for adrenal insufficiency may be increased by findings on routine electrolyte studies, glucose tests, and blood smears. Typical electrolyte changes due to the loss of cortisol and aldosterone include low serum sodium and high serum potassium levels. Elevation of serum levels of blood urea nitrogen and calcium may be observed in a significant number of patients. The elevation of serum calcium level is related to decreased renal excretion as well as increased bone resorption. Eosinophilia and leukocytosis can be found on blood smears. Hypomagnesemia and hypoglycemia may be seen in some patients. In acute crisis, these abnormalities may not have had time to develop.
Treatment of autoimmune thyroid disorder may cause unmasking of adrenal insufficiency and cause adrenal crisis.234, 235
Diagnosis
An 8.00am serum cortisol level of greater than 20 ug/dl excludes the disorder. A plasma ACTH of greater than 22 pmol/l is common with primary adrenocortical insufficiency.213, 227 Definitive diagnosis is made by the lack of adequate response of cortisol to ACTH stimulation. An ACTH stimulation test may be performed in unstressed, untreated patients by giving 0.25 mg synthetic ACTH intravenously or intramuscularly and showing an increase in cortisol in 1–2 hours.236 If the test does not show a rise in ACTH level, a more prolonged ACTH test given over a period of several days is required.
In primary adrenal insufficiency, the ACTH level will be elevated (>80 pg/ml), whereas in secondary adrenal insufficiency, it will be less than 20 pg/ml. An insulin tolerance test is used to diagnose secondary adrenal insufficiency since hypoglycemia causes stimulation of glucocorticoids via the hypothalamic–pituitary axis. Cortisol levels should rise dramatically in normal patients. An overnight metyrapone test can be used as an alternative. An oCRH testing can help to distinguish hypothalamic dysfunction from pituitary dysfunction. When 100 μg/kg intravenous oCRH is injected, patients with hypothalamic lesions have a greater increase in circulating ACTH levels than patients with pituitary lesions.213, 237
Once the diagnosis is established, tuberculosis must be excluded by a tuberculin skin test. If the result is negative, lack of anergy must be established. If the result of the skin test is positive, treatment is required.
A CT scan can be performed to differentiate between the etiologies of Addison's disease. Bilateral adrenal enlargement is the most common finding with tuberculous adrenalitis, but if the disease has been present for many years, shrunken fibrotic glands may be seen. Patients with autoimmune disease also may have small glands.238 When calcification is found, tuberculosis is strongly suggested, and neoplastic disease is excluded. The CT scan finding also may be suggestive of rare causes of the disorder, such as adrenal carcinoma, metastasis, and hemorrhage. MRI has no advantage over CT scanning in diagnosing these disorders.239
If no other etiology is found, idiopathic or autoimmune disease is diagnosed. Measurement of anti-adrenal antibodies is not necessary. Thyroid function tests should be performed because of the common association of hyperthyroidism. However, it is important not to initiate thyroid replacement therapy because glucocorticoid replacement alone may correct the abnormalities. This correction may occur either by inhibiting pituitary thyroid-stimulating hormone, stimulating thyroid hormone directly, or improving a generalized autoimmune dysfunction.240
Treatment
The usual glucocorticoid replacement therapy required for the patient with Addison's disease is the same as for CAH; 37.5 mg/day or its equivalent. Conventional practice is to administer two thirds of the total dose in the morning and one third in the afternoon or evening. Large doses given at bedtime may produce insomnia, and most patients find that a higher morning dose helps them to function. The dosage is adjusted to the lowest level that maintains weight and well being. It should be decreased if the patient has signs of iatrogenic Cushing's syndrome.
Fludrocortisone generally is used when mineralocorticoid replacement is required. The initial dosage is 0.05–0.20 mg/day. If edema, hypertension, or hypokalemia results, the dose should be decreased. If hypotension or hyperkalemia develops, the dose should be increased cautiously.
Prophylactic high-dose glucocorticoid replacement therapy is required for patients undergoing surgery or other stressful events; 300 mg intravenous cortisol should be given before the procedure, and 100 mg should be given every 6 hours for at least 24 hours. During pregnancy, the cortisol dose needs no adjustment unless nausea and vomiting prevent normal intake.241 With the onset of labor, 100 mg intravenous cortisol should be infused every 6–8 hours. This treatment is continued for 24 hours after delivery, when oral therapy is resumed.242
Perhaps the most critical factor in achieving optimal therapy is education of the patient. The patient must understand that she must take the replacement therapy daily and that she requires increased doses of glucocorticoid during periods of stress and illness. She must know to contact her physician if she has any illness that causes nausea and vomiting because it may prevent her from ingesting or absorbing her medication. She also should wear a bracelet or tag that identifies her diagnosis and treatment requirements, and she should carry commercially available prefilled syringes that contain a 4-mg ampule of dexamethasone or another glucocorticoid for emergency injections. It is best if the patient knows how to administer such injections.
Replacement with 25–50 mg of dehydroepiandrosterone has been studied and may improve quality of life.243, 244, 245
Adrenal transplantation to replace steroid therapy in patients with Addison's disease has been performed successfully in China, but no long-term follow-up has been published.246
Adrenal crisis
Acute adrenal crisis is a medical emergency. The patient has nausea, vomiting, severe hypotension, and dehydration precipitated by stress. It is more common in a patient with known Addison's disease as a result of intercurrent infection, trauma, or noncompliance. Blood for plasma cortisol determination should be obtained; however, 100 mg intravenous cortisol or its equivalent should be given immediately without awaiting results. This glucocorticoid treatment is life saving in patients with adrenal crisis, and there is no contraindication. A cortisol value of greater than 25 μg/dl excludes adrenal crisis. As reviewed by Phipps and Nagel in a previous version of this chapter, the clinical manifestations of acute adrenal insufficiency result from deficiencies of both aldosterone and cortisol. The patient is sodium depleted and hypovolemic. The assumption is made that the extracellular fluid volume is 20% depleted, which corresponds to approximately 5% of ideal body weight. Restoration of fluid is accomplished by infusing normal saline relatively rapidly; the first liter can be given over a period of 60 minutes. If present, hypoglycemia should be corrected by the addition of glucose to the intravenous fluid. Hyperkalemia may be present, and will resolve with glucocorticoid and fluid replacement therapy. A dose of 100 mg cortisol sodium succinate or a glucocorticoid equivalent should be administered intravenously every 6 hours. If the crisis was precipitated by severe concurrent illness or trauma, the high glucocorticoid doses used for initial replacement are maintained until the causal factor is controlled or eliminated.247
It is important to diagnose and provide appropriate treatment for a precipitating infectious process because the high doses of glucocorticoid used may mask signs of an infection and allow it to spread. Mineralocorticoid replacement therapy usually becomes necessary as the intravenous fluid and glucocorticoid replacement are tapered. After clinical recovery, the dose can be reduced by 50% daily until a replacement dose is achieved.
In patients who are being treated for acute adrenal insufficiency with no previous diagnosis, 4 mg dexamethasone sodium phosphate may be substituted for 100 mg cortisol during the initial glucocorticoid replacement therapy. This approach allows for simultaneous treatment and diagnostic testing with ACTH. Dexamethasone, unlike cortisol, has no mineralocorticoid activity. If the initial laboratory results show that the patient does not have adrenal insufficiency, steroid treatment should be withdrawn immediately to avoid the possibility of adrenal suppression.
ADRENAL TUMORS
The most frequent clinical presentation of a steroid-producing tumor of the adrenal cortex is Cushing's syndrome.248 Tumors also can cause syndromes of relatively pure virilization or feminization as well as hypoaldosteronism. Pure virilizing adrenal adenoma or carcinoma without symptoms of Cushing's syndrome may cause adrenal hyperandrogenism at any age.249, 250, 251, 252 Symptoms may develop over a period of 6 months to more than 10 years, and may include clitoromegaly, severe hirsutism, deepening of the voice, and amenorrhea. Circulating levels of T of greater than 2 ng/ml, androstenedione levels of greater than 380 ng/dl, and DHEAS levels of greater than 7000 ng/ml are detected in many, but not all, patients with adrenal androgen-producing tumors. Ultrasound, CT scans, and selective retrograde venous catheterization aid in the diagnosis.252, 253, 254 Treatment usually is surgical. The prognosis is good for benign adenoma, whereas the prognosis is poor for carcinoma.
REFERENCES
Adashi EY, Levin PA: Pathophysiology and evaluation of adrenal hyperandrogenism. Semin Reprod Endocrinol 4: 155, 1986 |
|
Speiser PW, White PC: Congeinital adrenal hyperplasias. N Engl J Med 349: 776, 2003 |
|
Feuillan P, Pang S, Schurmeyer T et al: The hypothalamic-pituitary-adrenal axis in partial (late-onset) 21-hydroxylase deficiency. J Clin Endocrinol Metab 67: 154, 1988 |
|
Merke DB, Bornstein SR:Congenital adrenal hyperplasia. Lancet 365: 2125, 2005 |
|
New MI: Extnesive clinical experience Nonclassical 21-hydroxylase Deficiency. J Clin Endocrinol Metab 91: 4205, 2006 |
|
Wilkins L, Crigler JF Jr, Silverman SH: Further studies on the treatment of congenital adrenal hyperplasia with cortisone, part II. The effects of cortisone on sexual and somatic development, with a hypothesis concerning the mechanism of feminization. J Clin Endocrinol 12: 277, 1952 |
|
Jones HW Jr, Verkauf F, Verkauf BS: Surgical treatment in congenital adrenal hyperplasia. Obstet Gynecol 36: 1, 1970 |
|
Krone N, Dhir V, Ivison HE, Arlt W. Congenital adrenal hyperplasia and P450 oxidoreductase deficiency. Clin Endrocrinol 66:162, 2007 |
|
White P: Steroid 11beta-hydroxylase Deficiency and Related Disorders. Endocrinol Metab Clin 30: x,2001 |
|
White PC, New MI, Dupont B: Molecular cloning of steroid 21-hydroxylase. Ann NY Acad Sci 458: 277, 1985 |
|
Carroll MC, Katzman P, Alicot EM et al: Linkage map of the human major histocompatibility complex including the tumor necrosis factor genes. Proc Natl Acad Sci USA 84: 8535, 1987 |
|
Kochham L, Janssen S, Knorr D et al: HLA class I-, complement C4- and 21-hydroxylase probes in the genetic analysis of 21-hydroxylase deficiency. J Clin Chem Clin Biochem 28: 413, 1990 |
|
White PC, Grossberger D, Onufer BJ et al: Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA 82: 1089, 1985 |
|
White PC, New MI, Dupont B: Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 83: 5111, 1986 |
|
Werkmeister JW, New MI, Dupont B, White PC: Frequent deletion and duplication of the steroid 21-hydroxylase genes. Am J Hum Genet 39: 461, 1986 |
|
Higashi Y, Yoshioka H, Yamane M et al. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc Natl Acad Dci USA 83:2841, 1986 |
|
White PC, New MI, Dupont B: HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc Natl Acad Sci USA 81: 7505, 1984 |
|
Holler W, Scholz S, Knorr D et al: Genetic differences between the salt-wasting, simple virilizing, and nonclassical types of congenital adrenal hyperplasia. J Clin Endocrinol Metab 60: 757, 1985 |
|
Mornet E, Crete P, Kuttenn F et al: Distribution of deletions of seven point mutations on CYP21B genes in three clinical forms of steroid 21-hydroxylase deficiency. Am J Hum Gen 48: 79, 1991 |
|
White PC, Tusie-Lana MT, New MI, Speiser PW. Mutations in steroid 21-hydroxylase (CYP21). Hum Mutat 3:373, 1994 |
|
Sinnott P, Collier S, Costigan C et al: Genesis by meiotic unequal crossover of a de novo deletion that contributes to steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA 87: 2107, 1990 |
|
Haglund-Stengler B, Ritzen EM, Gustafsson J, Luthman H: Haplotypes of the steroid 21-hydroxylase gene region encoding mild steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA 88: 8352, 1991 |
|
Speiser PW, New MI, White PC: Molecular genetic analysis of nonclassic steroid 21-hydroxylase deficiency associated with HLA-B14,DR1. N Engl J Med 319: 19, 1988 |
|
Levine LS, Dupont B, Lorenzen F et al: Cryptic 21-hydroxylase deficiency in families of patients with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 51: 1316, 1980 |
|
Claahsen-van der Grinten HL, Sweep FC, Blickman JG et al: Prevalence of testicular adrenal rest tumours in male children with congenital. Eur J Endocrinol. 2007 Sep;157(3):339-44. |
|
Trinh L, Nimkarn S, New MI et al: Growth and pubertal characteristics in patients with congenital adrenal. J Pediatr Endocrinol Metab. 2007 Aug;20(8):883-91. |
|
Stikkelbroeck NM, Otten BJ, Pasci A, et als. High prevalence of testicular adrenal rest tumors, impaired spermatogenesis and leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J Con Endocrinol Metab 86:5721, 2001 |
|
Stikkelbroek NM, Suliman HM, Otten BJ et al. Testicular adrenal rest tumours in postpubertal males with congenital adrenal hyperplasie: sonographic and MR feature. Eur Radiol 13:1597, 2003 |
|
Pang S, Wallace MA, Hofman L et al: Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 81: 866, 1988 |
|
Rosenwaks Z, Lee PA, Jones GS et al: An attenuated form of congenital virilizing adrenal hyperplasia. J Clin Endocrinol Metab 49: 335, 1979 |
|
Hagenfeldt K, Janson PO, Holmdahl G et al: Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Reprod. 2008 Jul;23(7):1607-13. |
|
Chrousos GP, Loriaux DL, Mann DL, Cuter GB: Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med 96: 143, 1982 |
|
Benjamin F, Deutsch S, Saperstein H, Seltzer VL: Prevalence of and markers for the attenuated form of congenital adrenal hyperplasia and hyperprolactinemia masquerading as polycystic ovarian disease. Fertil Steril 46: 215, 1986 |
|
Escobar-Morreale HF, Sanchon R, San Milllian JL. A prospective study of the prevalence of nonclassical congenital adrenal hyperplasia among women presenting with hyperandrogenic symptoms and signs. J Clin Endocrinol Metab 93:527-533, 2008 |
|
Pang, S, Hotchkiss J, Drash AL et al: Microfilter paper method for 17a-progesterone radioimmunoassay: Its application for rapid screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab 45: 1003, 1977 |
|
Speiser PW, Dupont B, Rubinstein P et al: High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet 37: 650, 1985 |
|
Speiser PW: Prenatal and neonatal diagnosis and treatment of congenital adrenal hyperplasia. Horm Res. 2007;68 Suppl 5:90-2. |
|
van der Kamp HJ, Oudshoorn CG, Elvers BH et al: Cutoff levels of 17-alpha-hydroxyprogesterone in neonatal screening forcongenital adrenal hyperplasia should be based on gestational age rather than onbirth weight. J Clin Endocrinol Metab. 2005 Jul;90(7):3904-7. |
|
McKenna TJ, Jennings AS, Liddle GW, Burr IM: Pregnenolone, 17-OH-pregnenolone, and testosterone in plasma of patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 42: 918, 1976 |
|
Levine LS, Dupont B, Lorenzen F et al: Genetic and hormonal characterization of cryptic 21-hydroxylase deficiency. J Clin Endocrinol Metab 53: 1192, 1981 |
|
Milewicz A, Vescsei P, Korth-Schuta S et al: Development of plasma 21-deoxycortisol radioimmunoassay and application to the diagnosis of patients with 21-hydroxylase deficiency. J Steroid Biochem 21: 185, 1984 |
|
Lee P, Gareis FJ: Evidence for partial 21-hydroxylase deficiency among heterozygote carriers for congenital adrenal hyperplasia. J Clin Endocrinol Metab 41: 415, 1975 |
|
Gutai JP, Kowarski AA, Migeon CJ: The detection of the heterozygote carrier for congenital virilizing adrenal hyperplasia. J Pediatr 90: 924, 1977 |
|
Gutai JP, Lee PA, Johnsonbaugh RE et al: Detection of the heterozygous state in siblings of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Pediatr 94: 770, 1979 |
|
Lejeune-Lenain C, Cantraine F, Dufrasnes M et al: An improved method for the detection of heterozygosity of congenital virilizing adrenal hyperplasia. Clin Endocrinol 12: 525, 1980 |
|
Rosenfield RL, Helke J, Lucky AW: Dexamethasone preparation does not alter corticoid and androgen responses to adrenocorticotropin. J Clin Endocrinol Metab 60: 585, 1985 |
|
New MI, Lorenzen F, Lerner AJ et al: Genotyping steroid 21-hydroxylase deficiency: Hormonal reference data. J Clin Endocrinol Metab 57: 320, 1983 |
|
Peter M, Sippell WG, Lorenzen F et al: Improved test to identify heterozygotes for congenital adrenal hyperplasia without index case examination. Lancet 2: 1296, 1990 |
|
Dewailly D, Vantyghem-Haudiquet MC, Sainsard C et al: Clinical and biological phenotypes in late-onset 21-hydroxylase deficiency. J Clin Endocrinol Metab 63: 418, 1986 |
|
Azziz R, Zacur HA: 21-hydroxylase deficiency in female hyperandrogenism: Screening and diagnosis. J Clin Endocrinol Metab 69: 577, 1989 |
|
Siegel SF, Finegold DN, Lanes R et al: ACTH stimulation tests and plasma dehydroepiandrosterone sulfate levels in women with hirsutism. N Engl J Med 323: 849, 1990 |
|
Frasier SD, Thorneycroft IH, Weiss BA et al: Elevated amniotic fluid concentration of 17α-hydroxyprogesterone in congenital adrenal hyperplasia. J Pediatr 86: 310, 1975 |
|
Forest MG, Betuel H, Couillin P et al: Prenatal diagnosis of congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency by steroid analysis in the amniotic fluid of mid-pregnancy: Comparison with HLA typing in 17 pregnancies at risk for CAH. Prenat Diagn 1: 197, 1981 |
|
Pang S, Pollack MS, Loo M et al: Pitfalls of prenatal diagnosis of 21-hydroxylase deficiency congenital adrenal hyperplasia. Ann NY Acad Sci 458: 111, 1985 |
|
Gueux B, Fiet J, Couillin P et al: Prenatal diagnosis of 21-hydroxylase deficiency congenital adrenal hyperplasia by simultaneous radioimmunoassay of 21-deoxycortisol and 17-hydroxyprogesterone in amniotic fluid. J Clin Endocrinol Metab 66: 534, 1988 |
|
Forest MG, Betuel H, David M: Prenatal treatment in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Update 88 of the French multicentre study. Endocr Rev 15: 277, 1989 |
|
Mornet E, Boue J, Raux-Demay M et al: First trimester prenatal diagnosis of 21-hydroxylase deficiency by linkage analysis to HLA-DNA probes and by 17-hydroxyprogesterone determination. Hum Genet 73: 358, 1986 |
|
Couillin P, Boue J, Nicolas H et al: Prenatal diagnosis of congenital adrenal hyperplasia (21-OH-deficiency type) by HLA typing. Prenat Diagn 1: 25, 1981 |
|
Grosse-Wilde H, Valentine-Thon E, Passarge E et al: HLA-A,B,C,DR typing and 17-OHP determination for second trimester prenatal diagnosis of 21-hydroxylase deficient CAH. Prenat Diagn 8: 131, 1988 |
|
Higashi Y, Yoshioka H, Yamane M et al: Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: A pseudogene and a genuine gene. Proc Natl Acad Sci USA 83: 2841, 1986 |
|
Reindollar RH, Lewis JB, White PC et al: Prenatal diagnosis of 21-hydroxylase deficiency by the complementary deoxyribonucleic acid probe for cytochrome P450C-210H. Am J Obstet Gynecol 158: 545, 1988 |
|
Billerbeck AE, Mendonca BB, Pinto EM et als. Three novel mutations in CYP21 gene in Brazilian patients with the classical from of 21-hydroxylase deficiency due to a founder effect. J Clin Endocrinol Metab 87:4314, 2002 |
|
Wilson RC. Wei JQ, Cheng KC, Mercado AB, New MI. Rapid deoxyribonucleic acid analysis by allel-specific polymerase chain reaction for detection of mutations in steroid 21-hydroxylase gene. J Clin Endocrinol Metab 80: 1635, 1995. |
|
Oriola J, Plensa I, Manchuca I, et als. Rapid screening method for detecting mutations in the 21-hydroxylase gene. Clin Chem 43:557, 1997 |
|
Bongiovanni AM: Adrenogenital syndrome with deficiency of the 3-beta-hydroxysteroid dehydrogenase. J Clin Invest 41: 2086, 1962 |
|
Pang S: Congenital adrenal hyperplasia owing to 3 beta-hydroxysteroid dehydrogenase deficiency. Endocrinol Metab Clin North Am. 2001 Mar;30(1):81-99, vi-vii. |
|
Bongiovanni AM: Acquired adrenal hyperplasia: With special reference to 3&b.beta;-hydroxysteroid dehydrogenase. Fertil Steril 35: 599, 1981 |
|
Pang S, Lerner AL, Stoner E et al: Late-onset adrenal steroid 3&b.beta;-hydroxysteroid dehydrogenase deficiency, part I. A cause of hirsutism in pubertal and postpubertal women. J Clin Endocrinol Metab 60: 428, 1985 |
|
Lucky AW, Rosenfield RL, McGuire J et al: Adrenal androgen hyperresponsiveness to adrenocorticotropin in women with acne and/or hirsutism: Adrenal enzyme defects and exaggerated adrenarche. J Clin Endocrinol Metab 62: 840, 1986 |
|
Chang YT,Zhang L Alkaddour HS et al. Absence of molecular defect in the type 2 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene in premature pubarche children and hirsute female patients with moderately decreased adrenal 3beta- HSD activity. Pediatr Res 37;820, 1995. |
|
Berube D, Luu-The V, Lachance Y. Assignment of the human 3beta-hydroxysteroid dehydrogenase gene to the p13 band of chromosome 1 Cytogenet Cell Genet 52;199, 1989 |
|
Lorence MC, Corbin CJ, Kamimura N et al. Structural analysis of the gene encoding human 3 beta-hydroxysteroid dehydrogenase/delta 5-delta isomerase. Mol Endocrinol 4;1850, 1990 |
|
Rheaume E, Lachance Y, Zhao HF et al: Structure and expression of a new complementary DNA encoding the almost exclusive 3&b.beta;-hydroxysteroid dehydrogenase/&b.Delta;5 -&b.Delta;4 isomerase in human adrenals and gonads. Mol Endocrinol 5: 1148, 1991 |
|
Welzel M, Wustemann N, Simic-Schleicher G et al: Carboxyl-terminal mutations in 3beta-hydroxysteroid dehydrogenase type II cause J Clin Endocrinol Metab. 2008 Apr;93(4):1418-25. |
|
Simard J, Moisan AM, Morel Y: Congenital adrenal hyperplasia due to 3beta-hydroxysteroid Semin Reprod Med. 2002 Aug;20(3):255-76. |
|
Gallegos AJ, Berlineer DL: Transformation and conjugation of dehydroepiandrostenedione by human skin. J Clin Endocrinol Metab 27: 1214, 1967 |
|
Kim MH, Herrman WL: In vitro metabolism of dehydroepiandrosterone sulfate in foreskin, abdominal skin, and vaginal mucosa. J Clin Endocrinol Metab 28: 187, 1968 |
|
Thomas JP, Oake RJ: Androgen metabolism in the skin of hirsute women. J Clin Endocrinol Metab 38: 19, 1974 |
|
Lutfallah C, Wang W, Mason JI et al: Newly proposed hormonal criteria via genotypic proof for type II J Clin Endocrinol Metab. 2002 Jun;87(6):2611-22. |
|
Mermejo LM, Elias LL, Marui S et al: Refining hormonal diagnosis of type II 3beta-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2genotyping. J Clin Endocrinol Metab. 2005 Mar;90(3):1287-93. Epub 2004 Dec 7. |
|
Moran C, Potter HD, Reyna R et al: Prevalence of 3beta-hydroxysteroid dehydrogenase-deficient nonclassic adrenal hyperplasia in hyperandrogenic women with adrenal androgen excess. Am J Obstet Gynecol. 1999 Sep;181(3):596-600. |
|
Pang S, Carbunaru G, Haider A et al: Carriers for type II 3beta-hydroxysteroid dehydrogenase (HSD3B2) deficiency canonly be identified by HSD3B2 genotype study and not by hormone test. Clin Endocrinol (Oxf). 2003 Mar;58(3):323-31. |
|
Pang S, Levine LS, Lorenzen F et al: Hormonal studies in obligate heterozygotes and siblings of patients with 11&b.beta;-hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab 50: 586, 1980 |
|
Mornet E, Dupont J, Vitek A et al. Characterization of two genes encoding human steroid 11 beta-hydroxylase (p-450(11)beta). J Biol Chem 264:20961, 1989 |
|
Brautbar C, Roesler A, Landau H et al: No linkage between HLA and congenital adrenal hyperplasia due to 11&b.beta;-hydroxylase deficiency. N Engl J Med 300: 205, 1979 |
|
White PC, Dupont J, New MI et al: A mutation in CYP11B1 associated with steroid 11 &b.beta;-hydroxylase deficiency in Jews of Moroccan origin. J Clin Invest 87: 1664, 1991 |
|
Ulick S, Wang JZ, Morton DH. The biochemical phenotypes of two inborn errors in the biosynthesis of aldosterone. J Clin Endocrinol Metab 74:1415, 1992. |
|
Maroulis GB, Manlimos FS, Garza R, Abraham GE: Serum cortisol and 11-desoxycortisol levels in hirsute premenopausal women. Obstet Gynecol 4: 388, 1976 |
|
Rose LI, Newmark SP, Strauss JS: Adrenocortical hydroxylase deficiencies in acne vulgaris. J Invest Dermatol 66: 324, 1976 |
|
Newmark S, Dluhy RC, Williams GH et al: Partial 11- and 21-hydroxylase deficiencies in hirsute women. Am J Obstet Gynecol 127: 594, 1977 |
|
Toaff ME, Toaff R, Chayen R: Congenital adrenal hyperplasia caused by 11&b.beta;-hydroxylase deficiency with onset of symptoms after one spontaneous pregnancy. Am J Obstet Gynecol 121: 202, 1975 |
|
Zachmann M, Tassinari D, Prader A: Clinical and biochemical variability of congenital adrenal hyperplasia due to 11&b.beta;-hydroxylase deficiency: A study of 25 patients. J Clin Endocrinol 56: 222, 1983 |
|
Birnbaum MD, Rose LI: Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol 63: 445, 1984 |
|
Schumert Z, Rosenmann A, Landau H et al: 11-Deoxycortisol in amniotic fluid: Prenatal diagnosis of congenital adrenal hyperplasia due to 11&b.beta;-hydroxylase deficiency. Clin Endocrinol 12: 257, 1980 |
|
Rosler A, Weshler N, Leiberman E et al: 11&b.beta;-Hydroxylase deficiency congenital adrenal hyperplasia: Update of prenatal diagnosis. J Clin Endocrinol Metab 66: 830, 1988 |
|
Bhangoo A, Anhalt H, Ten S et al: Phenotypic variations in lipoid congenital adrenal hyperplasia. Pediatr Endocrinol Rev. 2006 Mar;3(3):258-71. |
|
Baker BY, Lin L, Kim CJ et al: Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab. 2006 Dec;91(12):4781-5 |
|
Fujieda K, Tajima T, Nakae J et al. Spontaneous puberty in 46XX subjects with congenital lipoid adrenal hyperplasia. Ovarian steroidogenesis is spared to some extent despite inactivating mutations in the steroidogenic acute regulatory protein (StAR) gene. J Clin Invest 99; 1265, 1997 |
|
Kim CJ, Lin L, Huang N et al: Severe combined adrenal and gonadal deficiency caused by novel mutations in thecholesterol side chain cleavage enzyme, P450scc. J Clin Endocrinol Metab. 2008 Mar;93(3):696-702. |
|
Sugawara T Holt JA, Driscoll D et al. Human steroidogenic acute regulatory protein functional activity in COS-1 cels, tissue specific expression, and mapping of the structural gene to 8p11.2 and a pseudogene to chromosome 13. Proc Natl Acad Sci USA 92:4778, 1995. |
|
Miller WL: StAR search--what we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol Endocrinol. 2007 Mar;21(3):589-601. |
|
Mantero F, Scarlini C: Enzymatic defects of steroidogenesis: 17α Hydroxylase. In New MI, Levine LS (eds): Adrenal Diseases in Childhood, Vol 13, Pediatric and Adolescent Endocrinology, p 83. New York, S Karger AG, 1984 |
|
Yanase T, Sanders D, Shibata A et al: Combined 17α-hydroxylase/17.20-lyase deficiency due to a 7-base pair duplication in the N-terminal region of the cytochrome P45017 alpha (CYP17) gene. J Clin Endocrinol Metab 70: 1325, 1990 |
|
Kenny FM, Malvau P, Migeon CJ: Cortisol production rate in newborn babies, older infants and children. Pediatrics 31: 360, 1963 |
|
Brook CGD: The management of classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol 33: 559, 1990 |
|
Kreiger DT, Allen W, Rizzo F, Kreiger HP: Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab 32: 266, 1971 |
|
Knorr D, Bidlingmaier F, Kuhle U: Diagnosis and monitoring of therapy of the various enzymatic defects causing congenital adrenal hyperplasia by semiautomatic capillary gas-liquid chromatography. Horm Res 16: 201, 1982 |
|
Meakin JW, Tantongco MS, Crabbe J et al: Pituitary-adrenal function following long-term steroid therapy. Am J Med 29: 459, 1960 |
|
Danowski TS, Bonessi JV, Sabeth G et al: Probabilities of pituitary-adrenal responsiveness after steroid therapy. Ann Intern Med 61: 11, 1964 |
|
Graber AL, Ney RL, Nicholson WE et al: Natural history of pituitary-adrenal recovery following long-term suppression with corticosteroids. J Clin Endocrinol Metab 25: 11, 1965 |
|
Fujieda K, Reyes FI, Blankstein J, Faiman C: Pituitary-adrenal function in women treated with low doses of prednisone. Am J Obstet Gynecol 137: 962, 1980 |
|
Boyers SP, Buster JE, Marshal JR: Hypothalamic-pituitary-adrenal cortical function during long-term low-dose dexamethasone therapy in hyperandrogenized women. Am J Obstet Gynecol 142: 330, 1982 |
|
Kehlet H, Binder C: Value of an ACTH test in assessing hypothalamic-pituitary-adrenocortisol function in glucocorticoid-treated patients. Br Med J 2: 147, 1973 |
|
Brook CGD: The management of classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (oxf) 33: 559, 1990 |
|
Lin-Su K, Vogiatzi MG, Marshall I et al: Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2005 Jun;90(6):3318-25. |
|
Volkl TM, Simm D, Beier C et al: Obesity among children and adolescents with classic congenital adrenal.Pediatrics. 2006 Jan;117(1):e98-105. |
|
Klingensmith GJ, Carcia SC, Jones JW Jr: Glucocorticoid treatment of girls with congenital adrenal hyperplasia: Effects on height, sexual maturation and fertility. J Pediatr 90: 996, 1977 |
|
Hagenfeldt K, Janson PO, Holmdahl G et al: Fertility and pregnancy outcome in women with congenital adrenal hyperplasia. due Hum Reprod. 2008 Jul;23(7):1607-13. |
|
Moran C, Azziz R, Weintrob N, et al. Reproductive Outcome of Women with 21-hydroxylase deficient nonclassic adrenal hyperplasia. J Coin Endocrinol Metab 91:3451, 2006 |
|
Spritzer P, Billaud L, Thalabard JC et al: Cyproterone acetate versus hydrocortisone treatment in late-onset adrenal hyperplasia. J Clin Endocrinol Metab 70: 642, 1990 |
|
David M, Forest MG: Prenatal treatment of congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency. J Pediatr 105: 799, 1984 |
|
Evans MI, Chrousos GP, Mann DW et al: Pharmacologic suppression of the fetal adrenal gland in utero. JAMA 253: 1015, 1985 |
|
Chrousos GP, Evans MI, Loriaux DL et al: Prenatal therapy in congenital adrenal hyperplasia. Ann NY Acad Sci 458: 156, 1985 |
|
Speiser PW, Laforgia N, Kato K et al: First trimester prenatal treatment and molecular genetic diagnosis of congenital adrenal hyperplasia (21-hydroxylase deficiency). J Clin Endocrinol Metab 70: 838, 1990 |
|
Carpenter PC: Diagnostic evaluation of Cushing's syndrome. Endocrinol Metab Clin North Am 17: 445, 1988 |
|
Stewart PM: Adrenal cortex. In William's Textbook of Endocrinology, Tenth Edition , p 491-551. Philadelphia, PA: Saunders, 2003. |
|
Newell-Price J, Bertangna X, Grossman AB, Nierman LK. Cushing's Syndrome. Lancet 367:1605-1617, 2006. |
|
Van Cauter E, Refetoff S: Evidence for two subtypes of Cushing's disease based on the analysis of episodic cortisol secretion. N Engl J Med 312: 1343, 1985 |
|
Belsky JL, Cuello B, Swanson LW et al: Cushing's syndrome due to ectopic production of corticotropin-releasing factor. J Clin Endocrinol Metab 60: 496, 1985 |
|
Muller OA, Von Werder K. Ectopic production of ACTH and corticotropin-releasing hormone (CRH). J Steroid Biochem Mol Biol 43: 403-408, 1992. |
|
Marieb NJ, Spangler S, Kashgarian M et al: Cushing's syndrome secondary to ectopic cortisol production by an ovarian carcinoma. J Clin Endocrinol Metab 57: 737, 1983 |
|
Hermus AR, Pieters GF, Smals AG et al: Transition from pituitary dependent to adrenal-dependent Cushing's syndrome. N Engl J Med 318: 966, 1988 |
|
Van Berkhout FT, Croughs RJM, Wulffraat NM et al: Familial Cushing's syndrome due to nodular adrenocortical dysplasia is an inherited disease of immunological origin. Clin Endocrinol 31: 185, 1989 |
|
Ross E, Marshall-Jones P, Friedman M: Cushing's syndrome: Diagnostic criteria. Q J Med 35: 149, 1966 |
|
Plotz CM, Knowlton A, Raga C: The natural history of Cushing's syndrome. Am J Med 13: 597, 1952 |
|
Kasperlik-Zaluska AA, Migdalska BM, Zgliczynski S. Adrenocortical carcinoma: a clinical study and treatment results of 52 patients. Cancer, 75:2587-2591, 1995. |
|
Azzspardi J, William E: Pathology of nonendocrine tumors associated with Cushing's syndrome. Cancer 22: 274, 1968 |
|
Liddle P, Nicholson W, Island D et al: Clinical and laboratory studies of ectopic humoral syndromes. Recent Prog Horm Res 2: 283, 1969 |
|
Odagiri E, Yamanaka Y, Ishiwatari N et al: Studies on pituitary-gonadal function in patients with Cushing's syndrome. Endocrinol Jpn 35: 421, 1988 |
|
Bertagna C, Orth D: Clinical and laboratory findings and results of therapy in 58 patients with adrenocortical tumors admitted to a single medical center. Am J Med 71: 855, 1981 |
|
Gabrilove JL, Freiberg E, Nicolis GL: Peripheral blood steroid levels in Cushing's syndrome due to adrenocortical carcinoma or adenoma. Urology 22: 576, 1983 |
|
Ross EJ, Linch DC: Cushing's syndrome: Killing disease. Discriminating value of signs and symptoms aiding early diagnosis. Lancet 2: 646, 1982 |
|
Soffer L, Ianncerone A, Gabrilove J: Cushing's syndrome: A study of 50 patients. Am J Med 30: 129, 1961 |
|
Saruta T, Suzuki H, Handa M et al: Multiple factors contribute to the pathogenesis of hypertension in Cushing's syndrome. J Clin Endocrinol Metab 62: 575, 1986 |
|
Starkman MN, Schteingart DE: Neuropsychiatric manifestations of patients with Cushing's syndrome: Relationship to cortisol and adrenocorticotropic hormone levels. Arch Intern Med 141: 215, 1981 |
|
Graham BS, Tucker WS: Opportunistic infections in endogenous Cushing's syndrome. Ann Intern Med 101: 334, 1984 |
|
Findling JW, Raff H. Clinical Review: Clinical Review: Cushing's Syndrome: Important Issues in Diagnosis and Management. J Clin Endocrinol Metab 91:3746-3753, 2006. |
|
Nieman LK, Ilias I. Evaluation and treatment of Cushing's Syndrome. Am J Med 118: 1340-1346, 2005. |
|
Nieman LK, Biller BM, Findling JW et al: The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008 May;93(5):1526-40. |
|
Schteingart DE: Cushing's syndrome. Endocrinol Metab Clin North Am 18: 311, 1989 |
|
Melby JC: Assessment of adrenocortical function. N Engl J Med 285: 735, 1971 |
|
Parilatoo F, Smilo P, Forsham P: A rapid screening test for Cushing's syndrome. JAMA 113: 720, 1965 |
|
Carroll B, Curtis G, Mendelo J: Neuroendocrine regulation in depression. Arch Gen Psychiatry 33: 1039, 1976 |
|
Jubiz W, Meikle A, Levinson R: Effects of diphenylhydantoin on the metabolism of dexamethasone. N Engl J Med 283: 11, 1970 |
|
Liddle GW: Test of pituitary-adrenal suppressibility in the diagnosis of Cushing's syndrome. J Clin Endocrinol Metab 20: 1539, 1960 |
|
Besser G, Edwards G: Cushing's syndrome. J Clin Endocrinol Metab 1: 451, 1972 |
|
Cutler GB Jr: A Review of Endocrinology: Diagnosis and Treatment, p 565. Bethesda, MD, FAES, 1983 |
|
Bruno OD, Rossi MA, Contreras LN et al: Nocturnal high-dose dexamethasone suppression test in the aetiological diagnosis of Cushing's syndrome. Acta Endocrinol 109: 158, 1985 |
|
Biemond P, de Jong FH, Lamberts SWJ: Continuous dexamethasone infusion for seven hours in patients with the Cushing syndrome. Ann Intern Med 112: 738, 1990 |
|
Gambardella S, Tamburrano G, Giaccari A et al: Intravenous dexamethasone and subsequent ACTH test in comparison with dexamethasone oral test in the diagnosis of Cushing's syndrome: A report of 20 cases. J Endocrinol Invest 12: 163, 1989 |
|
Yanovski JA, Cutler GBJ, Chrousos GP, Niemaan LK. Corticotropin-releasing hormone stimulation following low-dose dexamethasone administration: a new test to distinguish Cushing's syndrome from pseudo-Cushing's states. JAMA 269:2232-2238, 1993. |
|
Buchfielder G, Paccotti P, Minetto M. The accuracy of CT and MR evaluation of the sella turcica for detection of adrenocorticotrphic hormone-secreting adenomas in Cushing's disease. Am J Neuroradiol 14:1183-1190, 1993. |
|
Chrousos GP, Schulte HM, Oldfield EH: The corticotropin-releasing factor stimulation test. N Engl J Med 310: 622, 1984 |
|
Nieman LK, Chrousos GP, Oldfield EH et al: The ovine corticotropin-releasing hormone stimulation test and the dexamethasone suppression test in the differential diagnosis of Cushing's syndrome. Ann Intern Med 105: 862, 1986 |
|
Muller OA, Stalla GK, von Werder K: CRH in Cushing's syndrome. Horm Metab Res [Suppl] 16: 51, 1987 |
|
Oldfield EH, Chrousos GP, Schulte HM et al: Preoperative lateralization of ACTH-secreting pituitary microadenomas by bilateral and simultaneous inferior petrosal venous sinus sampling. N Engl J Med 312: 100, 1985 |
|
Kaltsas GA, Giammulis MG, Newell-Price JD. A critical analysis of the value of simultaneous inferior petrosal sinus sampling in Cushing's disease and the occult ectpopic adrenocorticotropin syndrome. J Clin Endocrinol Metab 84:487-492, 1999. |
|
Vignati F, Berselli ME, Scialfa G et al: Bilateral and simultaneous venous sampling of inferior petrosal sinuses for ACTH and PRL determination: Preoperative localization of ACTH-secreting microadenomas. J Endocrinol Invest 12: 235, 1989 |
|
Landolt AM, Valavanis A, Girard J et al: Corticotrophin-releasing factor-test used with bilateral, simultaneous inferior petrosal sinus blood-sampling for the diagnosis of pituitary-dependent Cushing's disease. Clin Endocrinol 25: 687, 1986 |
|
Korobkin M, Francis IR. Adrenal Imaging. Semin Ultrasound CT MR 16:317-330, 1995. |
|
Doppman JL, Miller DL, Dwyer AJ et al: Macronodular adrenal hyperplasia in Cushing disease. Radiology 166: 347, 1988 |
|
McLeod MK, Thompson NW, Gross MD et al: Sub-clinical Cushing's syndrome in patients with adrenal gland incidentalomas. Am Surg 56: 398, 1990 |
|
Kazerooni EA, Sisson JC, Shapiro B et al: Diagnostic accuracy and pitfalls of [iodine-131] 6&b.beta;-iodomethyl-19-norcholesterol (NP-59) imaging. J Nucl Med 31: 526, 1990 |
|
Taylor L, Ayers JWT, Gross MD et al: Diagnostic considerations in virilization: Iodomethyl-norcholesterol scanning in the localization of androgen secreting tumors. Fertil Steril 46: 1005, 1986 |
|
Kumar R, David R, Sayle BA, Lamki L: Adrenal scintigraphy. Semin Roentgenol 23: 243, 1988 |
|
Fig LM, Gross MD, Shapiro B et al: Adrenal localization in the adrenocorticotropic hormone-independent Cushing syndrome. Ann Intern Med 109: 547, 1988 |
|
Sarkar SD, Cohen EL, Beierwaltes WH et al: A new and superior adrenal imaging agent, 131 I-68, 3A-iodomethyl-19-norcholesterol (NP-59): Evaluation in humans. J Clin Endocrinol Metab 45: 353, 1977 |
|
Hoefnagel CA. Metaiodobenzylguanidine and somatostatin in oncology: role in the management of neural cest tumours. Eur J Nucl Med 21:561-581, 1994. |
|
Pacak K, Ilias I, Chen CC et al: The role of [(18)F]fluorodeoxyglucose positron emission tomography and syndrome. J Clin Endocrinol Metab. 2004 May;89(5):2214-21. |
|
Biller BM, Grossman AB, Stewart PM et al: Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2008 Jul;93(7):2454-62. |
|
Aron DC, Findling JW, Tyrrell JB: Cushing's disease. Endocrinol Metab Clin 16: 705, 1987 |
|
Mampalam TJ, Tyrrel B, Wilson CB: Transsphenoidal microsurgery for Cushing's disease: A report of 216 cases. Ann Intern Med 109: 487, 1988 |
|
Shimon I, Ram Z, Cohen ZR, Hadami M. Transphenoidal surgery for Cushing's disease: endocrinological follow-up monitoring of 82 patients. Neurosurgery 51:57-61, 2002. |
|
Howlett TA, Plowman PN, Wass JAH et al: Megavoltage pituitary irradiation in the management of Cushing's disease and Nelson's syndrome: Long-term follow-up. Clin Endocrinol 31: 309, 1989 |
|
Petrovich Z, Jozsef G, Yu C, Apuzzo ML. Radiotherapy and sterotactic radiosurgery for pituitary tumors. Neurosurg Clin N Am 14:1470166, 2003. |
|
Wells SA Jr. The role of laparoscopic surgery in adrenal disease. J Clin Endocrinol Metab 83:3041-3043, 1998. |
|
Perry RR, Nieman LK, Cutler GB et al: Primary adrenal causes of Cushing's syndrome. Ann Surg 210: 59, 1989 |
|
Allolio B, Hahner S, Weismann D, Fassnacht M. Management of adrenocortical carcinoma. Clin Endocrinol 60:273-287, 2004. |
|
Reincke M, Allolio, Kaulen D et al: The effect of sodium valporate in Cushing's disease, Nelson's syndrome and Addison's disease. Klin Wochenschr 66: 686, 1988 |
|
Invitti C, DeMartin M, Brunani A et al: Treatment of Cushing's syndrome with the long-acting somatostatin analogue SMS 201-995 (Sandostatin). Clin Endocrinol 32: 275, 1990 |
|
Arnaldi G, Polenta B Cardinaletti M, Boscaro M. Potential indications for somatostatin analogs in Cushing's syndrome. J Endocrinol Invest 28:106-110, 2005. |
|
Sonino N, Boscaro M, Falio F. Pharmocologic management of Cusing syndrome: new targets for therapy. Treat Endocrinol 4:87-94, 2005. |
|
Nieman LK. Medical therapy of Cushing's disease. Pituitary 5:77-82, 2002. |
|
Thoren M, Adamson U, Sjoberg HE: Aminoglutethimide and metyrapone in the management of Cushing's syndrome. Acta Endocrinol 109: 451, 1985 |
|
Nussey SS, Price P, Jenkins JS et al: The combined use of sodium valproate and metyrapone in the treatment of Cushing's syndrome. Clin Endocrinol 28: 373, 1988 |
|
Sonino N: Current trends in the endocrine use of ketoconazole. J Endocrinol Invest 11: 741, 1988 |
|
Higashi Y, Omura M, Suzuki K et al: Ketoconazole as a possible universal inhibitor of cytochrome P-450 dependent enzymes: Its mode of inhibition. Endocrinol Jpn 34: 105, 1987 |
|
Burrin JM, Yeo TH, Ashby MJ et al: Effect of ketoconazole on adrenocorticotrophic hormone secretion in vitro and in vivo. J Endocrinol 108: 37, 1986 |
|
Boscaro M, Sonino N, Rampazzo A et al: Response of pituitary-adrenal axis to corticotrophin releasing hormone in patients with Cushing's disease before and after ketoconazole treatment. Clin Endocrinol 27: 461, 1987 |
|
Stalla GK, Stalla J, Huber M et al: Ketoconazole inhibits corticotropic cell function in vitro. Endocrinology 122: 628, 1988 |
|
Loose DS, Stover P, Feldman D: Ketoconazole binds to glucocorticoid receptors and exhibits glucocorticoid antagonist activity in cultured cells. J Clin Invest 72: 404, 1983 |
|
Weber MM, Luppa P, Engelhardt D: Inhibition of human adrenal androgen secretion by ketoconazole. Klin Wochenschr 67: 707, 1989 |
|
Martikainen H, Heikkinen J, Ruokonen A, Kauppila A: Hormonal and clinical effects of ketoconazole in hirsute women. J Clin Endocrinol Metab 66: 987, 1988 |
|
Engelhardt D, Jacob K, Doerr HG: Different therapeutic efficacy of ketoconazole in patients with Cushing's syndrome. Klin Wochenschr 67: 241, 1989 |
|
Angeli A, Frairia R: Ketoconazole therapy in Cushing's disease. Lancet 1: 821, 1985 |
|
McCance DR, Hadden DR, Sheridan KB et al: Clinical experience with ketoconazole as a therapy for patients with Cushing's syndrome. Clin Endocrinol 27: 593, 1987 |
|
Loli P, Berselli ME, Tagliaferri M: Use of ketoconazole in the treatment of Cushing's syndrome. J Clin Endocrinol Metab 63: 1365, 1986 |
|
Amado JA, Pesquera C, Gonazalez EM et al: Successful treatment with ketoconazole of Cushing's syndrome in pregnancy. Postgrad Med J 66: 221, 1990 |
|
Lewis JH, Zimmerman HJ, Benson GD et al: Hepatic injury associated with ketoconazole therapy. Gastroenterology 86: 503, 1984 |
|
Lake-Bakaar G, Scheuer PJ, Sherlock S: Hepatic reactions associated with ketoconazole in the United Kingdom. Br Med J 294: 419, 1987 |
|
Benini F, Savarin T, Senna GE et al: Diagnostic and therapeutic problems in a case of adrenal tuberculosis and acute Addison's disease. J Endocrinol Invest 13: 597, 1990 |
|
Arlt W Allolio B. Adrenal Insufficiency. Lancet 361:1881-1893, 2003 |
|
Rosenthal FD, Davies MK, Burden AC: Malignant disease presenting as Addison's disease. Br Med J 17: 1591, 1979 |
|
Kung AWC, Pun KK, Lam K et al: Addisonian crisis as presenting feature in malignancies. Cancer 65: 177, 1990 |
|
Rosenfield RL, Rich BH, Wolfsdorf JI et al: Pubertal presentation of congenital &b.Delta; 5-3&b.beta;-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 51: 345, 1980 |
|
Seidenwurm DJ, Elmer EB, Kaplan LM et al: Metastases to the adrenal glands and the development of Addison's disease. Cancer 54: 552, 1984 |
|
Bigazzi PE: Autoimmunity of the adrenals. In Volpe R (ed): Autoimmunity and Endocrine Disease, p 109. New York, Marcel Dekker, 1985 |
|
Nerup J: Addison's disease: Serological studies. Acta Endocrinol 76: 142, 1974 |
|
Khoury EL, Hammond L, Bottazzo GF et al: Surface-reactive antibodies to human adrenal cells in Addison's disease. Clin Exp Immunol 45: 48, 1981 |
|
Nagamine K, Peterson P Scott HS , et al. Positional cloning of the APECED gene. Nat Genet 17;393-398, 1997 |
|
Leor J, Levartowsky D, Sharon C: Polyglandular autoimmune syndrome, type 2. South Med J 82: 374, 1989 |
|
Schatz DA, Winter WE: Autoimmune polyglandular syndrome. II: Clinical syndrome and treatment. Endocrinol Metab Clin North Am. 2002 Jun;31(2):339-52. |
|
Trence DL, Morley JE, Handwerger BS: Polyglandular autoimmune syndromes. Am J Med 77: 107, 1984 |
|
Elder M, Maclaren N, Riley W: Gonadal autoantibodies in patients with hypogonadism and/or Addison's disease. J Clin Endocrinol Metab 52: 1137, 1981 |
|
Neufield M, Maclaren N, Blizzard R: Two types of autoimmune Addison's disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine 60: 355, 1981 |
|
Bouillon R: Acute adrenal insufficiency. Endocrinol Metab Clin North Am. 2006 Dec;35(4):767-75, ix. |
|
Falorni A, Laureti S, De Bellis A et al. Italian Addison Network Study; Update of Diagnostic Criteria for the Etiological Classification of Primary Adrenal Insufficiency. J Clin Endocrinol Metab 2004; 89:1598-1604. |
|
Moser HW, Mahmood A, Raymond GV: X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007 Mar;3(3):140-51. |
|
Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis adn therapy. Brain 120:1485-508, 1997 |
|
Moser J, Lutz Y, Stoeckel ME. The gene responsible for adrenoleukodystrophy encodes a peroxisomal membrane protein. Hum Mol Genet 3:265-271, 1994 |
|
Nerup J: Addison's disease: Clinical studies. A report of 108 cases. Acta Endocrinol 76: 127, 1974 |
|
Tobin MV, Aldridge SA, Morris AI et al: Gastrointestinal manifestations of Addison's disease. Am J Gastroenterol 84: 1302, 1989 |
|
Shaikh MG, Lewis P, Kirk JM: Thyroxine unmasks Addison's disease. Acta Paediatr. 2004 Dec;93(12):1663-5. |
|
Graves L 3rd, Klein RM, Walling AD: Addisonian crisis precipitated by thyroxine therapy: a complication of type 2autoimmune polyglandular syndrome. South Med J. 2003 Aug;96(8):824-7. |
|
Schulte HN, Chrousos GP, Argerinos P et al: The corticotropin-releasing hormone stimulation test: A possible aid in the evaluation of patients with adrenal insufficiency. J Clin Endocrinol Metab 58: 1064, 1983 |
|
Bethune JR: The diagnosis and treatment of adrenal insufficiency. In DeGroot L (ed): Endocrinology, Vol 2, pp 1647. Philadelphia, WB Saunders, 1989 |
|
Vita JA, Silverberg SJ, Goland RS et al: Clinical clues to the cause of Addison's disease. Am J Med 78: 461, 1985 |
|
Baker DE, Glazer GM, Francis IR: Adrenal magnetic resonance imaging in Addison's disease. Urol Radiol 9: 199, 1988 |
|
Farah DA, Boag D, Moran F, McIntosh S: High concentrations of thyroid-stimulating hormone in untreated glucocorticoid deficiency: Indication of primary hypothyroidism? Br Med J 285: 172, 1982 |
|
Albert E, Dalaker K, Jorde R, Berge LN: Addison's disease and pregnancy. Acta Obstet Gynecol Scand 68: 185, 1989 |
|
Gold WR: Acute adrenal insufficiency. In Queenan J (ed): Managing Ob/Gyn Emergencies, p 248. NJ, Medical Economics, 1983 |
|
Gurnell EM, Hunt PJ, Curran SE et al: Long-term DHEA replacement in primary adrenal insufficiency: a randomized,controlled trial. J Clin Endocrinol Metab. 2008 Feb;93(2):400-9. |
|
Arlt W, Callies F, van Vligmen JC, et al. Dehydroepiandrosterone replacement in women with adrenal insufficiency. N Engl J Med 1999;341:1013-1020 |
|
Johannson G, Burman P, Wiren I, et at. Low dose dehydroepiandrosterone affects behavior in hypopituitary androgen-deficient women: a placebo-controlled trial. J Clin Endocrinol Metab 2002;87:2046-2052. |
|
Yan ZB, Bing ZX, Yang WR, Long WL: A study of cadaveric fetal adrenal used for adrenal transplantation to treat Addison's disease: Thirteen cases reported. Trans Proc 22: 280, 1990 |
|
Waise A, Young RJ: Pitfalls in the management of acute adrenocortical insufficiency: Discussion paper. J R Soc Med 82: 741, 1989 |
|
Freeman DA: Steroid hormone-producing tumors in man. Endocr Rev 7: 204, 1986 |
|
Saez JM, Rivarola MA, Migeon CJ: Studies of androgens in patients with adrenocortical tumors. J Clin Endocrinol 27: 615, 1967 |
|
Kenny FM, Hashida Y, Askari HA et al: Virilizing tumors of the adrenal cortex. Am J Dis Child 115: 445, 1968 |
|
Granoff AB, Abraham GE: Peripheral and adrenal venous levels of steroids in a patient with virilizing adrenal adenoma. Obstet Gynecol 53: 111, 1979 |
|
Gabrilove JL, Seman A, Sobvet R et al: Virilizing adrenal adenoma with studies on the steroid content of the adrenal venous effluent and a review of the literature. Endocr Rev 2: 462, 1981 |
|
Surrey ES, de Zeigler D, Gambone JC, Judd HL: Preoperative localization of androgen-secreting tumors: Clinical, endocrinologic, and radiologic evaluation of ten patients. Am J Obstet Gynecol 158: 1313, 1988 |
|
Friedman CI, Schmidt GE, Kim M, Powell J: Serum testosterone concentrations in the evaluation of androgen producing tumors. Am J Obstet Gynecol 153: 44, 1985 |