Molecular Genetics
Authors
INTRODUCTION
The field of medical genetics has undergone striking changes in recent years, primarily as a result of the application of recombinant DNA technology to clinical genetics. The potential for identifying and isolating human genes already has been realized; more than 1500 gene markers have been mapped to human chromosomes.1 Furthermore, the number of single-gene disorders potentially amenable to prenatal diagnosis is now unlimited. Thus, in no other area of clinical medicine has this technology had such an immediate and dramatic impact as it has in reproductive genetics.
ADVANTAGES OF DNA-BASED DIAGNOSES
Until recently, prenatal diagnosis of mendelian genetic disorders has been limited to conditions for which the biochemical defect was known and expressed in fetal tissues (e.g., amniotic fluid cells, chorionic villi, blood, skin, liver). For example, hemophilia A and sickle-cell anemia could be diagnosed only through fetal blood sampling and phenylketonuria (PKU) through fetal liver biopsies, procedures that confer considerable risk to the fetus. Disorders such as cystic fibrosis (CF) and Duchenne muscular dystrophy (DMD) were not detectable prenatally, because the biochemical basis was unknown. Of the more than 2000 known mendelian disorders,2 less than 100 are amenable to prenatal diagnosis using biochemical or morphological markers in fetal tissues.3 Now, however, through the use of recombinant DNA technology, in theory all mendelian disorders are amenable to prenatal diagnosis. In the following sections, the basic techniques used in molecular genetic diagnosis, examples of their application to prenatal detection of mendelian disorders, and the advantages and disadvantages of each approach will be discussed.
THE BASIC TECHNIQUES
Even if a gene is not expressed by a particular cell, virtually every cell contains the full complement of genes. For example, although genes-encoding hemoglobin is expressed only by reticulocytes, the DNA coding for hemoglobin is present in all cells, including amniotic fluid cells and chorionic villi. Furthermore, the precise biochemical or molecular defect need not be known in order to detect the DNA that codes for the mutant gene.
DNA-based analysis relies on the fact that there are virtually unlimited numbers of nucleotide-sequence differences in the DNA of different individuals. These differences can be detected by restriction fragment length polymorphisms (RFLPs), or RFLP analysis. Although most sequence differences have no pathologic significance, they can serve as markers for mutant genes that cause disease. RFLPs allow diagnosis of affected individuals in families with a known genetic disease, even if the exact defect in the gene (i.e., mutation) is unknown. In cases where the disease-causing mutation is known, RFLP analysis may be used (e.g., sickle cell anemia), or, alternatively, a variety of methods may be employed for direct mutation detection (e.g., cystic fibrosis).
DNA-based analyses of genetic disease utilizes the following technologies: restriction endonuclease (restriction enzyme) digestions, immobilization of DNA by Southern or dot blotting, separation of DNA fragments by electrophoresis, visualization by hybridization to cloned DNA probes, and amplification of DNA using the polymerase chain reaction (PCR). These techniques, applied in a variety of ways, provide powerful tools for the diagnosis of genetic disorders.
Restriction Fragment Length Polymorphisms
Restriction endonucleases are bacterial enzymes that recognize and cut double-stranded DNA at specific nucleotide sequences. More than 400 restriction enzymes have been identified. (Examples of restriction enzymes are listed in Table 1) The sequences of DNA recognized by specific restriction enzymes are called restriction sites. Restriction sites are randomly distributed throughout the genome sites and may be found within or surrounding any particular gene. However, because more than 95% of human DNA does not code for gene products, most restriction sites are, by chance, located within the noncoding portion of genes.
TABLE 1. Examples of Restriction Enzymes and Nucleotide Recognition Sites
Enzyme | Recognition Sequence |
PvuII | CAG^CTG |
EcoI | G^AATTC |
MstII | CC^TNAGG |
CvnI | CC^TNAGG |
MspI | C^CGG |
TaqI | T^CGA |
^ = cutting site; N = any nucleotide
The DNA between two restriction sites is called a restriction fragment, and the size of a restriction fragment is determined by the distance between two restriction sites. Thus, when DNA is digested with a restriction enzyme it is cut into many fragments of varying lengths. Because nucleotide sequences vary from person to person, individuals will vary with respect to the number of restriction sites and the size of restriction-fragment lengths (i.e., restriction-fragment lengths are polymorphic). For example, in the theoretical case depicted in Figure 1, individuals lacking the second Pvu II restriction site will have one restriction fragment 5000 base pairs (or 5 kilobase pairs [kbp]) long, whereas individuals with the second restriction site will have two fragments of 3 kb and 2 kb in length. Differences between individuals with respect to the lengths of fragments are referred to as RFLPs. A polymorphism refers to the occurrence in a population of two or more forms of a gene, the least common having a frequency of at least 1%. Classic examples of human genetic polymorphisms are the ABO blood groups, serum transferrin, or the red-cell enzyme G6PD. Unlike RFLPs, however, the number of antigen, serum, or red-cell polymorphisms are limited in the human genome to less than 50 loci. RFLPs, on the other hand, provide geneticists with a virtually unlimited source of genetic markers through which diseases can be traced in families.
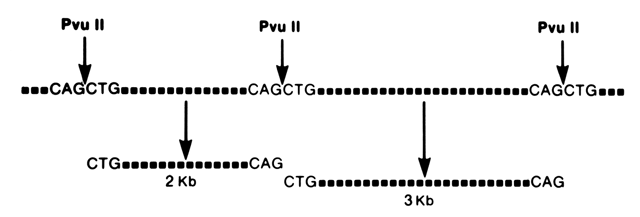{kind=link}
Detection of RFLPs by Electrophoresis, Southern Blotting, and Hybridization
When genomic DNA is digested with a particular restriction enzyme, fragments of many sizes result. The various size fragments can be physically separated on the basis of size by agarose gel electrophoresis (larger restriction fragments migrate through the gel more slowly than smaller restriction fragments). After electrophoresis, digested DNA appears under ultraviolet light as a smear (Fig. 2). To immobilize and preserve the DNA in the gel, the fragments are denatured (i.e., made single-stranded) and transferred to a membrane (such as nitrocellulose or charged nylon) by a method called “Southern blotting”4 that maintains the spatial orientation of the restriction fragments. Thus, the band patterns on the membrane are identical to those in the gel. To locate a particular gene within the many fragments on the blot, the membrane is soaked in a solution containing a radioactively (or enzymatically) labeled, single-stranded probe (i.e., DNA that is complementary to the gene of interest or to sequences that are located very close to {i.e., linked to} the gene of interest). The probe will hybridize (i.e., pair) to DNA that is complementary to it because both are single stranded. The radioactive Southern blot is then exposed to photographic film; fragments to which the probe hybridized are visualized as dark bands. These steps are illustrated in Figure 3.
{kind=link}
{kind=link}
Dot (or Slot) Blots
DNA can also be stabilized by “dotting” undigested DNA directly onto a membrane under vacuum. These dot, or “slot,” blots may be hybridized to a probe as described above. After autoradiography hybridization signals are viewed as a dark dot. Dot blots are most often used with PCR-amplified DNA and sequence-specific oligonucleotide (SSO) probes.5, 6
Polymerase Chain Reaction (PCR)
PCR is an in-vitro method for making multiple copies of specific DNA sequences.5 This powerful technique is capable of synthesizing over one million copies of specific DNA sequences in just a few hours. PCR allows diagnoses to be made on very small amounts of DNA (i.e., eliminating the need to culture cells to obtain larger amounts of DNA) and reduces the time required to make a diagnosis from a few weeks to a few days. Diagnoses can be made on amplified DNA either by direct visualization of the amplified product under ultraviolet light, or after hybridization to sequence-specific oligonucleotide probes. An oligonucleotide probe is a sequence of nucleotides (usually
DNA-BASED PRENATAL DIAGNOSIS
There are two general approaches to prenatal diagnosis through DNA analysis. The first, called the direct method, is the preferred method for prenatal diagnosis, but requires that the disease-causing mutation is known and detectable in a particular family. The second, called the indirect method, is based on linkage analysis and is more generally applicable, but less accurate than the direct method of diagnosis.
Direct Method Diagnosis
With this approach, DNA from the at-risk fetus is directly tested for the presence or absence of the abnormal gene. There are few potential sources of error with this method, provided the clinical diagnosis of the disease is correct. If more than one mutation results in a similar clinical phenotype (such as in families with cystic fibrosis and Duchenne muscular dystrophy), however, the direct test can be employed only if the specific mutation in that family is known. The applicability of the direct method is thus limited to genetic diseases in which the precise molecular defect is known. Although this is considered the ultimate goal for diagnosis of genetic disorders, at this time few genetic diseases can be so diagnosed.
Indirect Method of Diagnosis
This approach requires identifying RFLPs that are linked (lie within approximately 1000 kbp) to the disease gene. (RFLPs located this close to the gene will usually segregate with the gene, rather than undergo recombination at meiosis.) As discussed previously, RFLPs are randomly distributed throughout the genome. Thus, it is possible to identify RFLPs that demonstrate linkage to a disease in family studies, even if the abnormal gene itself has not been characterized. The presence of the RFLP can then be used to predict the presence of the abnormal gene in members of a family with affected individuals.
Despite the potential power of this approach, there are several limitations and sources of error. One requisite is that multiple family members, usually including at least one living affected relative, must be available. Also, costly and time-consuming studies must be performed in each at-risk family to determine whether the method will be applicable (i.e., informative) in the particular case. A third limitation is the possibility of genetic recombination in one of the parents' gametes between the disease gene and the linked marker. Because of this possibility, the accuracy of diagnosis using linked RFLPs is always less than 100%. The probability of recombination, however, is proportional to the chromosomal distance between the mutant gene and the RFLP. Thus, the smaller the distance between the restriction site and the disease gene, the more accurate the test. Whenever possible, several different linked RFLPs that map to either side of the defective gene should be used so that, if recombination occurs, it will be detected. The last potential source of error is false paternity. Obviously, if the biological father is not correctly identified in family studies, erroneous diagnoses may result.
Despite the above caveats, rapid progress in this area already has made possible the prenatal diagnosis of many important genetic diseases, using a combination of the laboratory techniques described above (Table 2). Given the rapidity of progress in this area, the prenatal diagnosis of many more mendelian disorders will become feasible in the not-too-distant future.
TABLE 2. Examples of Mendelian Disorders That Can Be Prenatally Diagnosed Using DNA-Based Diagnosis
Direct Method of Diagnosis
Sickle-cell anemia
α-thalassemia
β-thalassemia*
α-1, antitrypsin deficiency
Duchenne muscular dystrophy*
Cystic fibrosis*
Congenital adrenal hyperplasia*
Fragile X syndrome (X-linked mental retardation)
Indirect Method of Diagnosis (Linkage Studies)
Diseases for which the gene has been cloned, but all mutations are not detectable
β-thalassemia
Duchenne muscular dystrophy
Cystic fibrosis
Congenital adrenal hyperplasia
Diseases for which the gene has not been cloned
Huntington's disease
Adult-onset polycystic kidney disease (dominant form)
Myotonic dystrophy
Spinal muscular atrophy
* Available in families in whom exact mutation is known
Examples of how these techniques are used for both the direct and indirect diagnosis of sickle cell anemia, cystic fibrosis, and congenital adrenal hyperplasia due to 21-hydroxylase deficiency are described below. It should be noted that a variety of techniques may be used to diagnose these disorders; the following methods were chosen for illustrative purposes only.
EXAMPLES OF DNA-BASED DIAGNOSIS
Sickle Cell Anemia
Sickle cell anemia is an autosomal recessive disorder affecting one out of 400 African-American children in the United States. The defect results from a single nucleotide substitution (A to T) in the sixth codon (GAG to GTG) of the β-globin gene. This mutation leads to the amino acid substitution of valine for glutamine at position 6 of the β-globin polypeptide. Previously, diagnosis of sickle cell anemia was based on the detection of the abnormal type of hemoglobin in fetal blood. However, fetal blood sampling has become almost obsolete, as DNA-based diagnosis has developed.
The mutation causing sickle cell anemia coincidentally resides within the recognition sites of restriction enzymes Mst II, CvnI, and Bsu II. Individuals with the sickle cell mutation lack the restriction site present in individuals with normal β-globin genes. One procedure for diagnosing fetuses affected with sickle cell anemia using RFLP analysis is illustrated in Figure 4. Recently, more rapid diagnosis of sickle cell has been possible using PCR.6 With this method, a 725 bp region that includes the sickle cell mutation is amplified. The amplified DNA is digested with Bsu II, and then the digested product is separated by electrophoresis. The gel is stained with ethidium bromide, and the banding patterns can be directly visualized under ultraviolet light (Fig. 5). This procedure is much faster and more efficient because it eliminates the need to perform Southern blotting, hybridization with labeled probe, and autoradiography. This straightforward, direct method of diagnosis can be applied to any disease in which the mutation causing the disease removes or creates a restriction site.
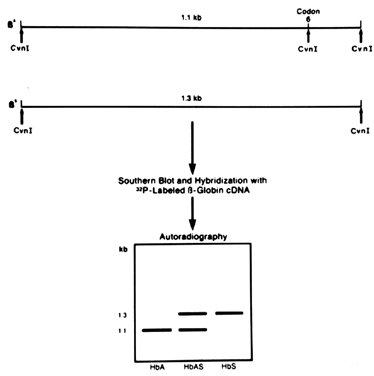{kind=link}
{kind=link}
Cystic Fibrosis
Cystic fibrosis (CF) is the most common autosomal recessive genetic disorder in the northern European population. Approximately one in 25 Caucasians are carriers (i.e., heterozygotes) for the gene, and one child in every 2500 births is affected with CF. Affected individuals rarely live past their thirties and suffer from debilitating pulmonary and digestive disorders throughout their lives. By demonstrating linkage between CF and RFLPs on chromosome 7, the CF gene was mapped to this chromosome in 1985,7, 8, 9, 10 but only recently was the CF gene itself identified and sequenced.11, 12, 13 A three-base pair deletion, resulting in the loss of a phenylalanine residue at position 508 (called delta F508), was found in 75% of CF chromosomes studied by Kerem and associates. The remainder of CF chromosomes each carry one of many (more than 100) less common mutations.
The direct detection of the delta F508 mutation does not require studying a living affected relative with CF and could be used to screen the general population for CF carrier status.13 Because only approximately 75% of CF carriers have the delta F508 mutation, however, 25% of carriers would go undetected if we screened for this mutation only. As a result, 25% of true carriers (or approximately 1% of individuals screened) will have a negative test, but will in truth be CF carriers. To further complicate matters, the frequency of the delta F508 mutation is lower in ethnic and racial groups other than northern European, non-Ashkenazi Caucasian populations. In these groups (such as African Americans, Ashkenazi Jews, southern and eastern Europeans), the frequency varies from 30% to 60%. Due to these limitations, screening for CF mutations is recommended currently only for relatives of individuals with CF and for spouses of known CF carriers. Screening for CF-carrier status in the general population is not recommended until additional mutations can be tested for, thereby reducing the false-positive rate and increasing the sensitivity of the screen.14, 15 It is anticipated that these problems will be overcome in the near future, and screening for CF carriers will become available at genetic centers.
Direct testing for delta F508 or other known mutations is useful in many families with a CF child. Because not all CF carriers have mutations that can be detected, however, linkage analysis using RFLPs is still required in some families. One method for detecting the delta F508 mutation utilizes PCR, dot blotting the amplified product, and hybridization to sequence-specific oligonucleotide (SSO) probes (Fig. 6).16 This method quickly determines whether an individual carries zero, one, or two copies of this mutation.
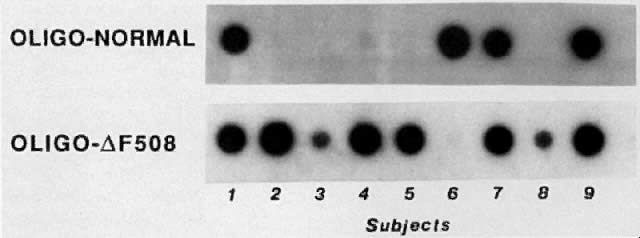{kind=link}
In CF families in which only one or neither parent carries a mutation that can be detected, prenatal diagnosis relies on family-based linkage studies. Figure 7A illustrates the relationship between the CF gene and a closely linked RFLP, KM. 19, in a family with three affected and two unaffected children. The pedigree of the family and RFLP banding patterns after electrophoresis of PCR-amplified DNA that was digested with the restriction enzyme, PstI, is shown in Figure 7B. The parents (I.1 and I.2) are presumed to be heterozygous carriers of the CF gene because they have three affected children. The affected children are assumed to be homozygous for the CF gene. The unaffected children can be either heterozygous carriers of the CF gene or can inherit normal genes from both parents and be homozygous normal. After DNA analysis, it is determined that both parents are heterozygous for the KM. 19 polymorphism (i.e., heterozygous for the presence of the restriction site; genotype +,-). The three affected children (II.1, II.3, II.5) are homozygous for the presence of the restriction site (genotype, +,+). Thus, we can deduce that each affected child inherited the chromosome containing the + allele from both parents, and the CF gene must be on these parental chromosomes. Both parental chromosomes with the - allele must therefore carry the normal gene. We can further deduce that the brother with genotype, -,- (II.2), inherited the normal gene from each parent and is not a carrier for CF. The sister with genotype +,- (II.4), inherited the chromosome with the normal gene from one parent and the chromosome with the CF gene from the other parent and is presumed to be a CF carrier.17
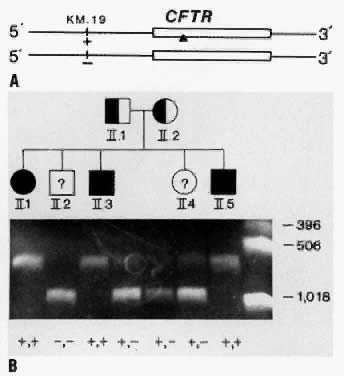{kind=link}
Prenatal diagnosis of CF in subsequent pregnancies in this family is also possible. Fetal DNA derived from chorionic villi or from amniotic fluid cells can be analyzed using the methods described above. As previously discussed, the major potential source of error in linkage studies is the probability of recombination between the restriction site and the abnormal gene in the parents' gametes. Thus, results from indirect DNA testing are given as a probability. For example, KM. 19 is within 100 kbp of the CF gene. Thus, the recombination frequency between the KM. 19 gene and the CF gene, determined from family studies, is small (less than 1%). If prenatal testing of a subsequent pregnancy in the couple in Figure 7 revealed the +,- genotype, the couple should be counseled that the probability that the fetus will have CF is equal to the probability that recombination occurred between the CF gene and the RFLP in the chromosome with the - allele (<1%). On the other hand, if the fetus was genotype, +,+, the probability that it will have CF is >99%. If the probability based on recombination calculations proves too uncertain, or for families in whom DNA diagnosis is not informative, measurement of amniotic fluid microvillar intestinal enzymes (i.e., alkaline phosphatase, γ-glutamyl transpeptidase, leucine amniopeptidase) may be useful, albeit not 100% sensitive or specific.18
Congenital Adrenal Hyperplasia (CAH)
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder of cortisol biosynthesis, which is caused in 95% of cases by a deficiency in the enzyme steroid 21-hydroxylase.19 This enzyme is required for the conversion of progesterone and 17-hydroxyprogesterone to 11-deoxycorticosterone and 11-deoxycorticosterol, which are intermediate products in mineralocorticoid and glucocorticoid biosynthesis, respectively. Due to a lack of feedback in CAH suppression of this pathway, there is a compensatory increase in ACTH, leading to adrenal hyperplasia and excessive secretion of precursor steroids. The increased secretion of adrenal androgens (such as DHEA and DHEAS) leads to increased conversion of these products to testosterone and dihydrotestosterone.
CAH occurs in a number of forms, ranging from mild to severe. The milder forms often present during childhood. More attenuated forms present at puberty (or even after puberty), with menstrual irregularities and infertility. The severe form is characterized by early virilization in utero, leading to marked masculinization of the external genitalia. Affected females may, in fact, be mistaken for males at birth, with bilateral cryptorchidism and hypospadias. The diagnosis may not be as obvious in males because the external genitalia are normal. Unrecognized salt wasting in neonates with CAH is often fatal because the inadequacy of glucocorticoids can lead to vascular collapse, shock, and death. The severe form of CAH occurs with a frequency of one in five to 10,000 births, and the milder (or attenuated) forms occur at a frequency of approximately one in 1000 births.20
The virilization process in utero begins early in pregnancy because the genital ridge forms at 9 to 10 weeks of gestation. Fortunately, the virilization process of female fetuses in utero can be inhibited by treatment of the mother during pregnancy with dexamethasone, thereby eliminating the need for extensive corrective surgery after birth. Because steroid treatment of the mother is not totally benign, however, treatment should be discontinued if the fetus is determined to be either homozygous or heterozygous for the normal 21-hydroxylase genes, or if the fetus is male. Thus, early and correct diagnosis of this genetic disease is of great importance. However, prenatal detection of CAH using biochemical tests for increased concentrations of amniotic fluid 17-hydroxyprogesterone are often inconclusive and are usually only feasible after 13 to 14 weeks' gestation.
The gene encoding the enzyme 21-hydroxylase has been mapped to the short arm of chromosome 6, positioned between the genes encoding HLA-B and HLA-DR.21, 22, 23 Gene probes for the 21-hydroxylase gene have been developed and molecular genetic studies of this region have demonstrated that two copies of the 21-hydroxylase gene (called A and B) are present in this region.23, 24 These two genes can be differentiated by hybridizing 21-hydroxylase gene probes to TaqI digested DNA: The A gene is detected on a 3.2 kb fragment, whereas the B gene resides on a 3.7 kb fragment. Detailed sequence analysis of the two genes has revealed that they are >90% homologous. Three deleterious mutations in the A gene render it nonfunctional, whereas the B gene is a functional gene, encoding the enzyme.25, 26 Analysis of southern blots of genomic DNA from patients with salt wasting CAH has revealed that, in about 25% of patients, the 21-hydroxylase B gene is either deleted or converted to the A gene.27 In these families, a direct diagnosis can be made by determining the presence or absence of the 3.7 kb fragment. In most cases, however, patients with CAH have the 3.7 kb TaqI fragment (B gene). In these families, the prenatal diagnosis of CAH depends on the indirect method, even though the biochemical defects for this disorder are known. RFLPs detected by probes for the HLA-B or HLA-DRβ loci have proven particularly useful for linkage studies in CAH families.
The author was supported in part by NIH research grant HD21244.
REFERENCES
Human Gene Mapping 10: Tenth International Workshop on Human Gene Mapping. Cytogenet Cell Genet 51:1, 1989 |
|
McKusick VA: Mendelian Inheritance in Man, 8th ed. Baltimore, John Hopkins Press, 1988 |
|
Simpson JL, Golbus MS, Martin AO, Sarto GE: Genetics in Obstetrics and Gynecology. New York, Grune & Stratton, 1982 |
|
Southern EM: Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98: 503, 1975 |
|
Erlich HA, Gelfand DH, Saiki RK: Specific DNA amplification. Nature 331: 461, 1988 |
|
Kazazian HH: Direct detection of point mutations. In Erlich HA (ed): PCR Technology: Principles and Applications for DNA Amplification. New York, Stockton Press, 1989 |
|
Tsui L-C, Buchwald M, Barker D et al: Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 230: 1054, 1985 |
|
White R, Woodward S, Leppert M et al: A closely linked genetic marker for cystic fibrosis. Nature 318: 382, 1985 |
|
Wainwright BJ, Scambler PJ, Schmidtke J et al: Localization of cystic fibrosis locus to human chromosome 7cen-q22. Nature 318: 384, 1985 |
|
Knowlton RG, Cohen-Haguenauer O, Van Cong N et al: A polymorphic DNA marker linked to cystic fibrosis is located on chromosome 7. Nature 318: 380, 1985 |
|
Rommens JM, Iannuzzi MC, Kerem B et al: Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 245: 1059, 1989 |
|
Riordan JR, Rommens JM, Kerem B-S et al: Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 245: 1066, 1989 |
|
Kerem B-S, Rommens JM, Buchanan JA et al: Identification of the cystic fibrosis gene: Genetic analysis. Science 245: 1073, 1989 |
|
Wilfond BS, Fost N: The cystic fibrosis gene: Medical and social implications for heterozygote detection. JAMA 263: 2777, 1990 |
|
Roberts L: CF screening delayed for awhile, perhaps forever. Science 247: 1296, 1990 |
|
Lemna WK, Feldman GL, Kerem B-S et al: Mutation analysis for heterozygote detection and the prenatal diagnosis of cystic fibrosis. N Engl J Med 322: 291, 1990 |
|
Feldman GL, Williamson R, Beaudet AL, O'Brien WE: Prenatal diagnosis of cystic fibrosis by DNA amplification for detection of KM-19 polymorphism. Lancet 2: 102, 1988 |
|
Brock DJH, Clarke HAK, Barron L: Prenatal diagnosis of cystic fibrosis by microvillar enzyme assay on a sequence of 258 pregnancies. Hum Genet 78: 271, 1988 |
|
Childs BM, Grumbach MM, Van Wyk JJ: Virilizing adrenal hyperplasia, a genetic and hormonal study. J Clin Invest 35: 213, 1962 |
|
Speiser PW, Dupont B, Rubinstein P et al: High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet 37: 650, 1985 |
|
Dupont B, Oberfield SE, Smithwick EM et al: Close genetic linkage between HLA and congenital adrenal hyperplasia (21-hydroxylase deficiency). Lancet 2: 1309, 1977 |
|
Dupont B, Virdis R, Lerner AJ et al: Distinct HLA-B antigen association for the salt-wasting and simple virilizing forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. In Terasaki PI (ed): Histocompatibility testing, p 660. Heidelberg, Springer, 1984 |
|
Carroll MC, Cambell RC, Porter RR: Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc Natl Acad Sci USA 82: 521, 1985 |
|
White PC, Grossberger D, Onufer BJ et al: Two genes encoding steroid 21-hydroxylase are located near the gene encoding the fourth component of complement in man. Proc Natl Acad Sci USA 82: 1089, 1985 |
|
White PC, New MI, Dupont B: Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 83: 5111, 1986 |
|
Higashi Y, Yoshioka H, Yamani M: Complete nucleotide sequences of two steroid 21-hydroxylase genes tandemly arranged in the human genome. Proc Natl Acad Sci USA 83: 2841, 1986 |
|
Werkmeister JW, New MI, Dupont B, White PC: Frequent deletion and duplication of the steroid 21-hydroxylase genes. Am J Hum Genet 39: 461, 1986 |