True Hermaphroditism
Authors
INTRODUCTION
True hermaphrodites (ovotesticular disorders of sex development) have ovarian as well as testicular tissue. The diagnosis has traditionally been applied only if an individual has (1) histologically verified ovarian follicles or proof of their prior existence (e.g., corpora albicantia) and (2) seminiferous tubules or spermatozoa. Fibrous stroma does not suffice in lieu of follicles, nor do Leydig cells suffice in lieu of tubules. The diagnosis is applied regardless of chromosome complement. True hermaphroditism has been reported in humans and in many animals; in some lower animals, in fact, hermaphroditism is functional. This review is, however, restricted to true hermaphroditism in humans. More extensive reviews are published elsewhere1, 2, with updates by the author.3, 4
HISTORICAL BACKGROUND AND DEFINITIONS
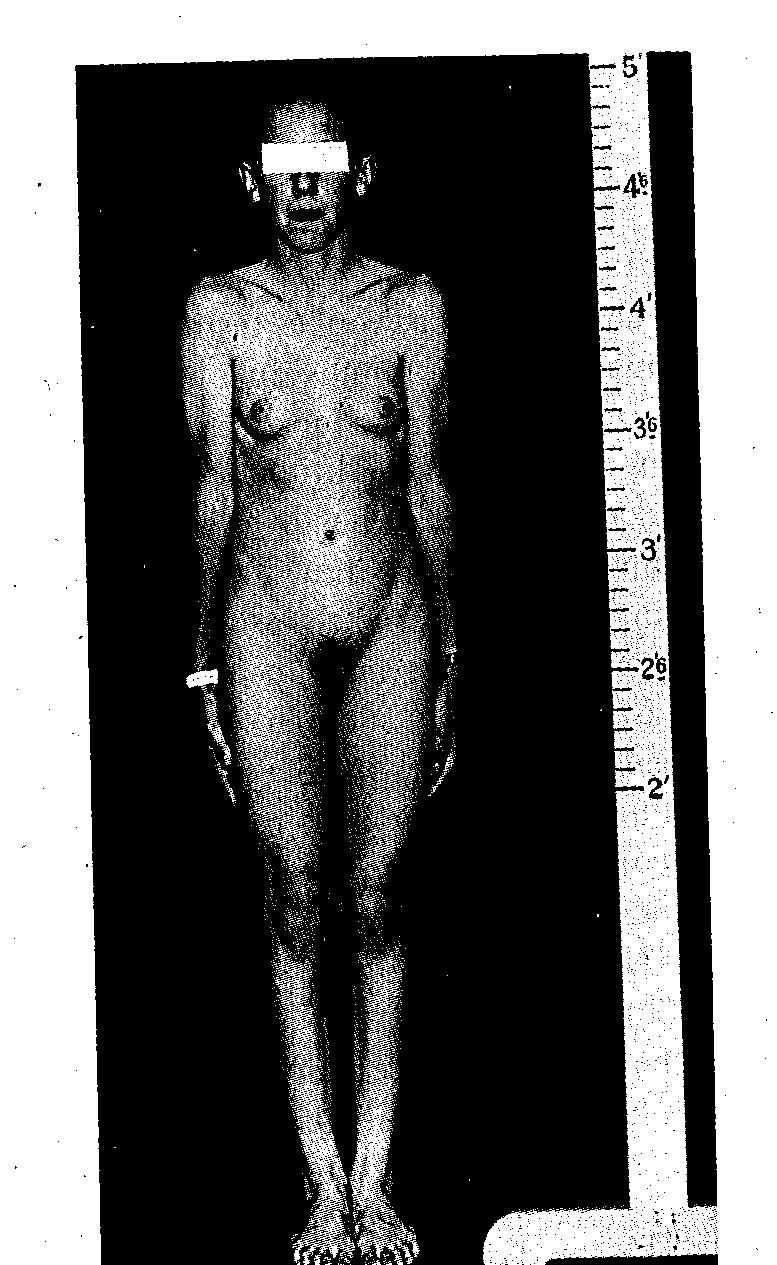
Klebs5 is usually credited with the first scientific consideration of true hermaphroditism. By the beginning of the modern cytogenetic era (circa 1958), approximately 200 true hermaphrodites had been documented. Analysis of these earlier cases led to certain clinical generalizations that still retain some validity. For example, most true hermaphrodites were raised as males, even though many had hypospadias and incomplete labioscrotal fusion. Gonads in these individuals consisted of (1) bilateral ovotestes (so-called bilateral gonadal hermaphroditism), (2) one ovary and one testis (alternating gonadal hermaphroditism), or (3) one ovotestis and a contralateral testis or ovary (unilateral gonadal hermaphroditism). Ovarian and testicular portions of ovotestes usually were juxtaposed end-to-end, facilitating diagnosis by inspection or palpation. A uterus was usually present. A fallopian tube was usually present ipsilateral to an ovary and often ipsilateral to an ovotestis. If a testis was present, a vas deferens and epididymis were usually present ipsilateral to the testis. At puberty, breast development usually occurred (Fig. 1) and menstruation often occurred.
Fig. 1. Photograph of an adolescent true hermaphrodite whose gonadal tissue is shown in Fig. 2. Breast development occurs in many true hermaphrodites, including paradoxically those reared as males. (From Sarto GE, Opitz JM, lnhorn ST. Consideration of sex chromosome abnormalities in man. In Benirschke K (ed): Comparative Mammalian Cytogenics. New York: Springer-Verlag, 1969.) |
{kind=link}
In 1959, Harnden and Armstrong6 and Hungerford et al.7 reported the first true hermaphrodites subjected to cytogenetic analysis. Both groups, surprisingly at the time, reported the karyotype to be 46,XX.
By 1977, cytogenetic studies had been performed on at least 240 live-born cases that fulfilled traditional histologic criteria of hermaphroditism.2 This furnished the basis for analyses of the author at that time. These clinical lessons remain generally applicable 30 years later.2 More recently, new nomenclature has been proposed for the disorders of sex development (DSD). In this classification, true hermaphroditism is termed ovotesticular DSD.
TYPES OF TRUE HERMAPHRODITISM (Ovotesticular disorders of sex development)
46,XX/46,XY
True hermaphroditism of the type 46,XX/46,XY is rarer than 46,XX true hermaphroditism, but about as common as the 46,XY type. Not all 46,XX/46,XY individuals are true hermaphrodites; some are phenotypically normal or have gonadal dysgenesis or male pseudohermaphroditism. In fact, fusion of XX and XY mouse zygotes usually produces not true hermaphroditism but, rather, clearly male or clearly female offspring.8
A 46,XX/46,XY chromosome complement could arise by (1) nondisjunction involving a 47,XXY or 46,XY zygote, with loss of certain cell lines and retention of others, or (2) chimerism.
The etiology of 46,XX/46,XY true hermaphroditism in humans is usually considered to be chimerism, rather than nondisjunction. Chimerism connotes the presence in a single individual of cells derived from different zygotes. The phenomenon is proved by detection of two populations of cells, usually erythrocytes with different blood types, in a single individual. At least three types of chimerism are known: (1) blood chimerism (interchange of blood cells between co-twins through placental anastomoses); (2) transplacental chimerism (interchange of fetal and maternal blood cells); and (3) whole-body chimerism. Whole-body chimerism, presumably the type of chimerism responsible for some 46,XX/46,XY true hermaphrodites, could result from: (1) fertilization of both an ovum and its polar body; (2) fertilization of each of two ova contained within a single binucleated follicle; (3) fertilization of ova derived from different follicles, followed by fusion; or (4) other related phenomena. Whole-body chimerism can be assumed if two or more genotypes are (1) present in nonhematogenous tissues (skin or gonads) or (2) persist in hematogenous tissues. The frequency of whole-body chimerism may be underestimated, since chimeric individuals are usually ascertained because of abnormal sexual development (e.g., true hermaphroditism). Thus, almost all recognized whole-body chimeras are heterosexual, although one might expect an equal number of isosexual chimeras (ascertainment bias). That whole-body chimerism has been detected in phenotypically normal individuals confirms that a 46,XX/46,XY karyotype is not invariably associated with true hermaphroditism.
Ascertainment of 46,XX/46,XY true hermaphrodites has been accomplished by studies restricted to lymphocytes as well as by studies of both lymphocytes and other tissues (e.g., skin or gonadal fibroblasts). Cytogenetic studies limited to lymphocytes have sometimes failed to detect more than one line. If additional tissues are studied, the minority cell line is most likely to be detected in gonadal fibroblasts and relatively less likely to be detected in skin fibroblasts. In addition, the proportion of 46,XY cells in lymphocytes bears no ostensible relationship to gonadal status. That is, a relatively high proportion of 46,XY cells in lymphocytes does not necessarily indicate that the gonadal tissue is predominantly testicular, nor, conversely, does a high proportion of 46,XX cells indicate that gonadal tissue is predominantly ovarian. The clinical significance of these observations is that analysis of multiple tissues is the only way to detect certain 46,XX/46,XY true hermaphrodites, although many can be detected by analyzing lymphocytes alone.
The external genitalia of known 46,XX/46,XY true hermaphrodites are usually either ambiguous or sufficiently masculinized to suggest to attending physicians that the sex of rearing should be male. A female sex of rearing was chosen in only three of 13 cases in one series.2 Even if reared as males, affected individuals usually show perineal or penoscrotal hypospadias and incomplete labioscrotal fusion.
The distribution of gonadal tissue is shown in Table 1. No single type predominates. Although the chromosome constitution of gonadal tissue has been determined in relatively few cases, 46,XY cells appear more likely to be present in a testis or ovotestis than in an ovary. Additional studies, however, are necessary to confirm this hypothesis. In aggregate, the right gonad is much more likely to contain testicular tissue than the left – a characteristic that 46,XX/46,XY true hermaphrodites share with other true hermaphrodites. There is no obvious explanation for the predilection for testicular development to take place on the right, but it is interesting to note that in many species, the right gonad is vestigial. Furthermore, following extirpation of the left ovary from newborn hens, the right gonad differentiates into a testis.9
Table 1. Clinical characteristics of true hermaphrodites with various chromosomal complements
Gonadal distribution (left:right) | ||||||||||||
Chromosomal complement (total cases)* | Sex of rearing (female/total)† | Uterus present‡ | Unicornuate or bicornuate uterus§ | OT:OT | O:T | T:O | O:OT | OT:O | T:OT | OT:T | Other | Total informative cases |
46,XX/46,XY (28) | 3/13 | 20/22 | 1/20 | 6 | 7 | 1 | 4 | 4 | 1 | 2 | 0 | 24 |
46,XX/47,XXY (13) | 3/10 | 8/10 | 2/8 | 4 | 2 | 3 | 1 | 0 | 0 | 0 | 2 | 12 |
45,X/46,XY (11) | 3/8 | 5/5 | 0/5 | 0 | 4 | 1 | 0 | 2 | 0 | 1 | 0 | 8 |
46,XY (37) | 2/17 | 17/19 | 2/17 | 0 | 12 | 4 | 4 | 0 | 4 | 2 | 2 | 28 |
Familial 46,XX (8)** | 1/10 | 2/10 | 0/2 | 6 | 0 | 0 | 3 | 0 | 1 | 0 | 0 | 10 |
Nonfamilial 46,XX (131) | 34/89 | 90/108 | 18/90 | 23 | 24 | 8 | 36 | 9 | 2 | 10 | 5 | 117 |
O, ovary; T, testis; OT, ovotestis.
*Complete descriptions not available for all cases.
†Includes only those cases in which attending physicians assigned a sex of rearing.
‡Includes cases in which uterus described as rudimentary
§Probably an underestimate; descriptions of uteri often incomplete.
**References 10, 11, 12, 13, 14, 15; cases of Kasdan et al.16 and Berger et al.17 not included.
From Simpson JL: True hermaphroditism: Etiology and phenotypic considerations. Birth Defects 14(6C): 9, 1978.
PHENOTYPE OF 46,XX TRUE HERMAPHRODITES
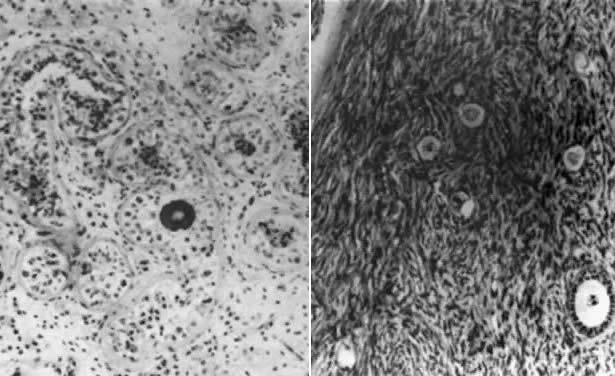
The ovarian and testicular portions of an ovotestis are usually juxtaposed end-to-end. By definition, both seminiferous tubules and ovarian follicles are present. Testicular tissue, whether existing as a separate testis or as one component of an ovotestis, is characterized by relatively few normal germ cells. The tubules are usually hyalinized and composed only of Sertoli cells (Fig. 2); Leydig cells may be hyperplastic. Although spermatozoa are rare, one possible 46,XX/46,XY case18 was fertile and had a sperm count of 20 × 106/ml. By contrast, ovarian tissue often contains numerous primordial follicles in various developmental stages. That ovarian tissue is often normal is also evidenced by breast development, cyclic menses, and histologic evidence of ovulation. However, histologic criteria for diagnosis introduce some biases favoring presence of normal oocytes in true hermaphrodites.
{kind=link}
Wiersma found that of 125 cases in South Africa reported in 2010, no patient showed normal male or female external genetalia.19 All except two were raised as males. Only nine of 125 showed separate urethral and vaginal openings. Paradoxically, breast development was typical at puberty, even with predominantly male external genitalia. This may have reflected atrophy of testicular tissue due to intraabdominal location. As noted, gonads may consist of two ovotestes; one ovary and one contralateral testis; or one ovotestis and a contralateral ovary or testis. Gonadal tissue may be located in the ovarian, inguinal, or labioscrotal regions. A testis or an ovotestis is more likely to be present on the right than on the left. Spermatozoa are rarely present;1 however, ostensibly normal oocytes may exist even in ovotestes. Gonads were palpable in only 59 of the 125 South African cases of Wiersma et al.,19, 20 and of these 59 only 17 bilateral. In 66 cases, gonads were in the pelvis. Ovotestes are the predominant gonadal form in South Africa and these were almost always pelvic in location. In the series of Wiersma tissue distribution was typically bipolar (ovarian cranial and testicular caudal), albeit with interdigitation. In 89% of ovotestes studied by Wiersma a mantle of ovarian tissue surrounded a core containing stroma and intermingled ovarian and testicular tissue. The clinical significance of gonadal intermingling is that extirpating gonadal tissue of a single type is not simple surgically.
Van Niekerk, whose elegant book in 1974 analyzed a presumably different South African cohort, reported at the time that the greater the proportion of testicular tissue in an ovotestis, the greater the likelihood of gonadal descent. In 80% of his cases, the testicular and ovarian components of ovotestes were juxtaposed end to end21 seemingly different from the recent experience of Wiersma. Van Niekerk collected cases from the region surrounding Pretoria, whereas Wiersma19, 20 is active in Durban. Thus, different tribal ancestry may exist. Van Niekerk noted that ovotestis may be detectable by inspection or palpation; testicular tissue is softer and darker than ovarian tissue. Ultrasound or magnetic resonance imaging (MRI) are now obligatory if the undesired portion of an ovotestis is to be extirpated.
Gonadal neoplasia and breast carcinoma have been reported.2, 22, 23 Gonadal neoplasia may reflect risks associated with intra-abdominal location of testicular tissue. This is consistent with data of Kuhnle and coworkers24 who estimated that neoplasia arises in 25% of 46,XY cases but in only 3% of 46,XX cases. The preponderance of gonads existing in the form of ovotestes in South Africa is less pronounced elsewhere. Vilain25 stated that the distribution worldwide is 34% ovotestes/ovary, 29% ovotestes/ovotestes and 25% ovary/testes.
A uterus is usually (90%) present based on cases reported worldwide and was as well in Van Niekerk’s sample.21 Sometimes the uterus is bicornuate or unicornuate.2 However, in his series, also from South Africa, Wiersma reported that only 73 of 125 had a “uterine structure”. Absence of a uterine horn usually indicates ipsilateral testis or ovotestis. The fimbriated end of the fallopian tube is often occluded ipsilateral to an ovotestis. Squamous metaplasia of the endocervix may occur.21 Menstruation is common and may be manifested as cyclic hematuria. Presence of a uterus in true hermaphroditism is diagnostically useful, particularly in the rare 46,XY case. Of individuals with genital ambiguity having a Y chromosome, only 46,XY hermaphrodites and 45,X/46,XY mosaics have a uterus.
Rarely is the uterus absent, although a few authors comment that uterine development was rudimentary or “merely a remnant”. Menstruation occurred in three of seven pubertal individuals with a uterus.2 The presence or absence of fallopian tubes or Wolffian derivatives reflects the ipsilateral gonad. A fallopian tube is invariably present ipsilateral to an ovary, whereas a vas deferens, epididymis, and, often, a seminal vesicle are usually present: ipsilateral to a testis. Either fallopian tubes or Wolffian derivatives may be present on the side of an ovotestis, although most often a fallopian tube is present. Although fallopian tubes and Wolffian derivatives are usually not both present on the same side, even on the side of an ovotestis, this combination has been observed occasionally.
PHENOTYPE OF 46,XX/47,XXY
True hermaphroditism is rarely observed associated with a 46,XX/47,XXY chromosome complement probably resulting from nondisjunction involving a 46,XY or 47,XXY zygote. The karyotype can be explained readily by postulating loss of certain cell lines and retention of others. The possibility of chimerism has not been excluded, however, or even, in most cases, vigorously pursued.
Of 13 cases detected up to 1977, most were studied by cultured lymphocytes. The sex of rearing was chosen by the attending physicians in ten cases. The gonadal distribution shows no obvious pattern (Table 1). A uterus was present in eight of ten cases, two being unicornuate. Prevalences of bicornuate and unicornuate uteri seem likely to be underestimated, since the uterus was often not completely described. The occurrence of unicornuate or bicornuate uteri would be predicted because androgens and the Müllerian inhibitory factor (MIF) influence embryonic ductal differentiation through local diffusion from the fetal testes.
PHENOTYPE OF 45,X/46,XY
Individuals with a 45,X/46,XY karyotype display a spectrum of phenotypes, ranging from almost normal males to females indistinguishable from those with 45,X gonadal dysgenesis and the Turner stigmata.1 The different phenotypes presumably reflect different tissue distributions of 45,X and 46,XY cells. This assumption, however, has not been proved and, in fact, often cannot be demonstrated, although one should recall that in gonadal cultures fibroblasts rather than germ cells are cultured.
The phenotype most often associated with 45,X/46,XY mosaicism is mixed or asymmetric gonadal dysgenesis, characterized by a unilateral streak gonad and a contralateral testis. In most 45,X/46,XY individuals, no ovarian follicles are detectable, and hence the diagnosis of true hermaphroditism is inappropriate.
Although 11 45,X/46,XY true hermaphrodites have been reported, only eight were completely described. A male sex of rearing was chosen in five of the eight cases. A uterus was found in five cases. Five of eight had one ovary and one testis, and in four of these five, the testis was on the right. No case had bilateral ovotestes (Table 1).
Phenotype of other mosaic or chimeric true hermaphrodites
True hermaphroditism has been associated with various other chromosomal abnormalities, including 45,X/46,XX; 46,XX/47,XYY; 46,XX/46,XY/ 47,XXY; 46,XX/47,XXY/49,XXYYY; 45,X/46,XY/47,XYY; and 46,XX/48,XXYY. These karyotypes probably arise by mitotic nondisjunction, although chimerism has not been excluded. Because only a few cases have been associated with a given karyotype, generalizations would be unwise; however, alternating gonadal hermaphroditism occurs relatively frequently.
PHENOTYPE OF 46,XY TRUE HERMPAHRODITES
About 40 cases of 46,XY true hermaphroditism have been reported, although a complete description is not available for all of them. Over half were Japanese; 46,XY true hermaphroditism appears to be the most common type of true hermaphroditism in Japan, in contrast to its relative rarity outside Asia.
A sex of rearing was assigned in 19 cases, and in only two cases was the female role chosen. This indicates that the external genitalia are more masculine in appearance than in 46,XX/46,XY true hermaphroditism, a suggestion consistent with published descriptions of external genitalia. Quantitation of genital virilization, however, is difficult. The gonadal distribution in Table 1 shows a high frequency of the alternating type, specifically a left ovary and a right testis. No 46,XY true hermaphrodites definitely had bilateral ovotestes, although one possible case was reported by Sandberg et al.26 A uterus was present in 17 of 19 cases for which complete descriptions were available; two uteri were unicornuate and one was bicornuate. Development of Wolffian and Müllerian derivatives was similar to that in other true hermaphrodites.
At least two 46,XY true hermaphrodites27, 28 developed gonadoblastomas, suggesting that 46,XY true hermaphrodites show the same predilection for neoplastic transformation as do individuals with 45,X/46,XY mosaicism or XY gonadal dysgenesis. Breast carcinoma has also been reported in two true hermaphrodites; one was 46,XX21 and the karyotype of the other29 is unknown.
ETIOLOGY OF TRUE HERMAPHRODITES
46,XX/46,XY true hermaphroditism is rare and usually caused by chimerism. The presumptive explanation is chimerism, the presence in a single individual of two or more cell lines by definition each derived from different zygotes. Some 46,XY cases may also be unrecognized chimeras.30 46,XX/47,XXY individuals are probably more likely to have resulted from nondisjunction or anaphase lag. In 46,XX true hermaphrodites, possible explanations include translocation during paternal meiosis of SRY from the Y to an X; translocation of SRY from the Y to an autosome; or activation (depression) of autosomal genes. The former can be excluded as uncommon because very few 46,XX cases receive DNA sequences from their father’s Y.31 Rarity of X-Y interchange in true hermaphrodites contrasts with its frequent occurrence in 46,XX men, 80% of whom show SRY as a result of such an interchange. A few 46,XX ovotesticular cases do show a mutant SRY, and somatic mutation can result in gonadal mosaicism associated with true hermaphroditism.32 Autosomal genes seem to be a more likely explanation for 46,XX true hermaphroditism. This hypothesis is supported by the existence of families characterized by either multiple siblings with XX true hermaphroditism or families in which both 46,XX males and 46,XX true hermaphrodites occur in the same kindred. [For references, see prior Section on 46,XX Males (XX Sex Reversal)]. In these kindreds, the 46,XX men usually show genital ambiguity, unlike the typical 46,XX male who shows normal male external genitalia. Familial XX true hermaphrodites seem more likely to be characterized by bilateral ovotestes and absence of the uterus than nonfamilial true hermaphroditism;2 both gonads in familial true hermaphroditism tend to be morphologically similar and ovotestes. This suggests a central basis for gonadal perturbation.2 Given that differing gonadal composition per side logically should more likely be the result of chimerism. Derepression of a normally dormant autosomal gene conferring male differentiation could provide an explanation for unscheduled (testicular) gonadal development in 46,XX individuals. We have already observed instructive cases which resulted in 46,XX sex reversal (female genotype to male phenotype) in which duplication of SOX9 has not been well studied in true hermaphrodites causes of 46,XX sex reversal in the absence of SRY.
REFERENCES
Simpson JL: Disorders of Sexual Differentiation: Etiology and Clinical Delineation. New York: Academic Press, 1976 |
|
Simpson JL: True hermaphroditism: etiology and phenotypic considerations. Birth Defects Orig Artic Ser. 1978;14(6C):9-35. |
|
Simpson JL: Disorders of the gonads, genital tract, and genitalia. (In) Principles and Practice of Medical Genetics., 5th ed. (D.L. Rimoin, J.M. Connor, R.E. Pyertiz, B.R. Korf eds.). Churchill-Livingstone, New York, pp. 2055-2092, 2007. |
|
Simpson JL: Disorders of sexual differentiation. (In) Pediatric and Adolescent Gynecology, 3rd ed. (J.S. Sanfilippo, ed). W.B. Saunders Company, New York, NY, in press. |
|
Klebs E: Handbuch der pathologischen Anatomie. Berlin: Hirschwald, 1876 |
|
Harnden DG, Armstrong CN: The chromosomes of a true hermaphrodite. Br Med J 2: 1287, 1959 |
|
Hungerford DA, Donnelly A J, Nowell PC, Beck S: The chromosome constitution of a human phenotypic intersex. Am J Hum Genet 11: 215, 1959 |
|
Mystkowska ET, Tarkowski AK: Observations on CBA-P/CBA-T6 T6 mouse chimeras. J Embryol Exp Morphol 20: 33, 1968 |
|
Jost A: Gonadal hormones in the sex differentiation of the mammalian fetus. In De Hann RL, Urspring H (eds): Organogenesis. New York: Holt, Rinehart & Winston, 1965 |
|
Clayton GW, Smith JD, Rosenberg HS: Familial true hermaphroditism in pre- and postpubertal genetic females. Hormonal and morphologic studies. J Clin Endocrinol Metab 18: 1349, 1958 |
|
Rosenberg HS, Clayton GW, Hsu TC: Familial true hermaphroditism. J Clin Endocrinol Metab 23: 203, 1963 |
|
Mori Y, Mizutani S: Familial true hermaphroditism in genetic females. Nippon Hinyokika Gakkai Zasshi (Jpn J Urol) 59: 857, 1968 |
|
Armendares S, Salamanca F, Cantu JM, del Castillo V, Nava S, Dominquez de la Piedra E, Cortes-Gallegos V, Gallegos A, Cervantes C, Parra A: Familial true hermaphroditism in three siblings: Clinical, cytogenetic, histological, and hormonal studies. Humangenetik 29: 99, 1975 |
|
Gallegos AJ, Guizar E, Armendares S, Cortes-Gallegos V, Cervantes C, Bedolla N, Parra S: Familial true hermaphroditism in three siblings: Plasma hormonal profiles and in vitro steroid biosynthesis in gonadal structures. J Clin Endocrinol Metab 42: 653, 1976 |
|
Fraccaro M, Tiepolo L, Zuffardi O, Chiumello O, Di Natale B, Gargantini L, Wolf U: Familial XX true hermaphroditism and the H-Y antigen. Hum Genet 48: 45, 1979 |
|
Kasdan R, Nankin HR, Troen P, Wald N, Pan S, Yanaihara T: Paternal transmission of maleness in human beings. N Engl J Med 288: 539, 1973 |
|
Berger R, Abonyi D, Nodot A, Vialatte J, Lejeune J: Hermaphrodisme vrai et “Garcon XX” dans une fratrie. Rev Europ Etud Clin Biol 15: 330, 1970 |
|
Shannon R, Nicolaides MJ: True hermaphroditism with oogenesis and spermatogenesis. Aust NZ J Obstet Gynacol 13: 84, 1973 |
|
Wiersma R: The clinical spectrum and treatment of ovotesticular disorder of sexual development. Adv Exp Med Biol. 2011;707:101-3. |
|
Wiersma R: True hermaphroditism in southern Africa: the clinical picture. Pediatr Surg Int. 2004 May;20(5):363-8. Epub 2004 May 26. |
|
Van Niekerk WA (1974) True Hermaphroditism: Clinical, Morphologic, and Cytogenetic Aspets. Harper & Row, Hagerstown |
|
Simpson JL, Photopulos G: Letter: Bilateral teratoma of testis in 2 brothers with 47,XXY Klinefelter'ssyndrome. Clin Genet. 1976 Mar;9(3):380-1. |
|
Verp MS, Harrison HH, Ober C et al: Chimerism as the etiology of a 46,XX/46,XY fertile true hermaphrodite. Fertil Steril. 1992 Feb;57(2):346-9. |
|
Kuhnle U, Schwarz HP, Lohrs U et al: Familial true hermaphroditism: paternal and maternal transmission of truesequences. Hum Genet. 1993 Dec;92(6):571-6. |
|
Vilain E: The genetics of ovotesticular disorders of sex development. Adv Exp Med Biol. 2011;707:105-6. |
|
Sandberg AA, Koepf GF, Crosswhite LH, Hauschka TS: The chromosome constitution of human marrow in various developmental and blood disorders. Am J Human Genet 12: 231, 1960 |
|
Park IJ, Pyeatte JC, Jones HW Jr, Woodruff JD: Gonadoblastoma in a true hermaphrodite with 46,XY genotype. Obstet Gynecol 40: 466, 1972 |
|
Szokol M, Kondrai G, Papp Z: Gonadal malignancy and 46,XY karyotype in a true hermaphrodite. Obstet Gynecol 49: 358, 1977 |
|
Moriarty JD: True hermaphroditism: Report of a case with mammary carcinoma. Am J Pathol 20: 799, 1961 |
|
Mikati MA, Najjar SS, Sahli IF et al: Microcephaly, hypergonadotropic hypogonadism, short stature, and minor anomalies:a new syndrome. Am J Med Genet. 1985 Nov;22(3):599-608. |
|
Ramsay M, Bernstein R, Zwane E et al: XX true hermaphroditism in southern African blacks: an enigma of primary sexualdifferentiation. Am J Hum Genet. 1988 Jul;43(1):4-13. |
|
Braun A, Kammerer S, Cleve H et al: True hermaphroditism in a 46,XY individual, caused by a postzygotic somatic pointhistological findings in a sporadic case. Am J Hum Genet. 1993 Mar;52(3):578-85. |