Genetics of Uterine Leiomyomas
Authors
INTRODUCTION
Leiomyomas of the uterus, also known as uterine fibroids or fibromas, are the most common neoplasms of the female genital tract. Although these benign smooth muscle tumors may develop at various sites within the body, they most frequently affect uterine myometrium. Leiomyomas constitute a major health issue for women, accounting for approximately 30% of all hysterectomies performed in the United States annually.1 From a study of serial sections of uteri, it has been estimated that up to 77% of women of reproductive age have one or more fibroids.2 Most affected women have multiple tumors, with the average number of tumors per uterus estimated to be 6.5.2 The biological sequelae of fibroids manifest as a spectrum of clinical symptoms. Some fibroids cause excessive menorrhagia, severe abdominal pain, urinary incontinence, and constipation; others develop as subclinical (asymptomatic) pelvic masses. Reproductive issues are also a concern, because fibroids are associated with infertility and, if present during a pregnancy, may contribute to spontaneous abortion, premature labor, or dystocia. Rarely, if ever, does a leiomyoma progress to a malignant leiomyosarcoma; such a clinical course is estimated at less than 0.1%. Although hysterectomy is frequently the treatment endpoint, one option is myomectomy, a more conservative surgical procedure that removes fibroids while preserving the uterus. This is a particularly attractive option for women who have not yet completed childbearing, but as many as 25% of women treated with myomectomies experience a recurrence of their tumors and ultimately undergo a hysterectomy for abatement of their symptoms.3
Leiomyomas are classified according to their location within the uterus. The most common type, the intramural fibroid, is located within the smooth muscle of the uterine wall and results in an irregular increase in fundal size. As a fibroid enlarges, it may expand toward the uterine surface, becoming subserous, and it may even infarct if it becomes pedunculated. In contrast, submucosal tumors can grow into the uterine cavity, causing attenuation of the endometrium, and they are most often associated with many of the typical symptoms described earlier. Less common is the polypoid form of a fibroid, which grows along a stalklike structure and can, at times, extend past the cervix into the vagina.4
An understanding of the development and pathogenesis of leiomyomas is the goal of much current research. Not surprising for a tissue so intimately connected with the action of hormones, data have accumulated suggesting the contributory roles of estrogen and progesterone. Generally, estrogen is believed to provide a major growth stimulus for fibroids, and it is fairly common for these tumors to shrink in size after menopause. In fact, certain treatment modalities target a reduction in estrogen levels by blocking gonadotropin release. Studies have also shown that progesterone plays an important role in fibroid development.5, 6
Although a significant number of studies have focused on identifying the specific genetic lesions in leiomyomas, evidence exists also for an inherited component in their etiology. Results of studies of twins and affected sister pairs, as well as the analysis of patients with multiple hereditary uterocutaneous leiomyomas, support a role for genetic factors in the development of these tumors.7, 8, 9, 10 The goal of this chapter on the genetics of leiomyomas is to summarize the current understanding of cytogenetic aberrations and gene expression with respect to their contributions to the pathogenesis of these tumors.
NONRANDOM CYTOGENETIC CHANGES OF LEIOMYOMAS
Attempts to categorize leiomyomas further have been aided by cytogenetic analyses of resected samples. Approximately 25–40% of fibroids show karyotypically detectable chromosomal abnormalities that are both nonrandom and tumor-specific.11, 12 These samples have been classified into several cytogenetic categories based on the chromosome aberration present and include the following subgroups: t(12;14)(q14-q15;q23–24), del(7)(q22q32), rearrangements involving 6p21, 10q, trisomy 12, and deletions of 1p3q. Although no relationship between patient age or parity and the type of chromosomal abnormality has been identified, the presence of a cytogenetic abnormality has been correlated with the anatomic location of a uterine fibroid.13 In this study, submucous fibroids were consistently shown to have fewer cytogenetic abnormalities (12%) compared with intramural (35%) and subserous (29%) fibroids. These data complement earlier work correlating increased tumor size with presence or type of chromosomal abnormalities.11, 14, 15
Clonality Studies of Uterine Leiomyomas
Cytogenetic analyses of multiple fibroids from a single uterus have demonstrated that the tumors can harbor different chromosomal changes, suggesting that each fibroid develops independently. X-inactivation studies, which exploit the phenomenon of Lyonization, have clearly demonstrated that fibroids develop as clonal lesions.16, 17, 18, 19 Initially, X-linked glucose-6-phosphate dehydrogenase (G6PD) isoenzyme analysis was used to demonstrate the independent clonal origin of multiple tumors within a single uterus.18, 19 However, a low degree of G6PD isoenzyme polymorphism among most females across various ethnic groups limited the usefulness of this analysis. Another approach, based on the CAG repeat polymorphism in the X-linked androgen receptor gene, has been used also to examine clonality.16 Experiments in this study demonstrated a random pattern of inactivation among multiple tumors: individual tumors exclusively expressed one or the other allele, confirming the hypothesis that multiple tumors within a single uterus arise independently. It is possible, however, that some leiomyomas develop from a common precursor cell; the discovery of identical cytogenetic abnormalities in multiple fibroids from the same patient argues for this path of development.11 Yet these changes may simply be representative of neoplastic smooth muscle. A study of a single patient with two independent fibroids, each showing different patterns of X-chromosome inactivation but identical del(7)(q21.2q31.2) derivative chromosomes, could be interpreted to support this latter hypothesis or reflect a stochastic occurrence of del(7)(q21.2q31.2).20
Studies of tumors that initially appeared to be polyclonal have shown that two independent fibroids can actually develop in extremely close proximity to each other.21 The discovery of heterogeneity of chromosomal aberrations is consistent with the multistep hypothesis of tumor development, in which the function (or dysfunction) of several genes at multiple loci results in fibroid growth. Abnormalities at several loci have been documented in individual tumors, and this heterogeneity may explain clinicopathologic differences seen in fibroids, including variation in size or response to hormonal treatments. With such heterogeneity it is possible that, rather than a required order to the mutation of critical genes, the combined action of individual genes and accumulation of mutated loci contribute to the growth of leiomyomas. This phenomenon has been studied and described in detail for a number of malignant tumors.22, 23
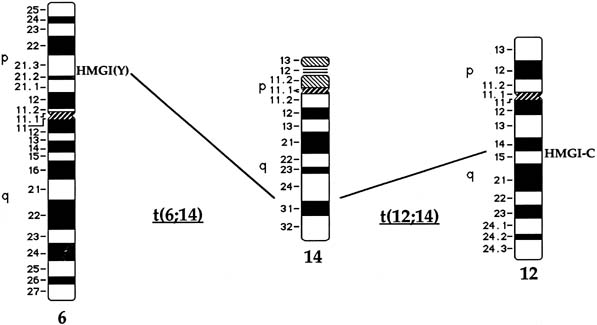
The t(12;14) Subgroup
Approximately 20% of karyotypically abnormal leiomyomas show a characteristic t(12;14) rearrangement, the most commonly observed chromosomal translocation in fibroids as well as the first cytogenetic anomaly to be associated specifically with uterine leiomyomas.24 The finding of rearrangements involving the same region of 12q in other mesenchymal solid tumors (e.g. angiomyxomas, breast fibroadenomas, endometrial polyps, hemangiopericytomas, lipomas, pulmonary chondroid hamartomas, salivary gland adenomas) supported the notion that a critical gene(s) for the process of tumorigenesis mapped to 12q14–q15 (Fig. 1).25, 26, 27, 28, 29, 30, 31, 32, 33 The fact that t(12;14) was frequently the sole detectable chromosomal alteration in a subset of fibroids lent additional support to the importance of genes mapping to the breakpoints involved in this translocation. After many efforts to map the critical region on chromosome 12, a single yeast artificial chromosome (YAC) clone was found to span cytogenetic breakpoints in uterine leiomyoma, pulmonary chondroid hamartoma, and lipoma specimens with t(12;14) translocations, and therefore was likely to include the critical gene.34 The HMGA2 [formerly designated as HMGIC] gene was first considered a positional candidate because it mapped within this YAC clone and eventually was identified as the disrupted gene in a series of mesenchymal tumors.34, 35 A candidate gene on chromosome 14 that may be involved in t(12;14) is RAD51L1, which has been identified as a partner of HMGA2.36, 37 RAD51L1 is a member of the recA/RAD51 recombination repair gene family. However, it is important to note that RAD51L1 spans a large genomic region of approximately 700 kilobases that potentially contains multiple other genes or regulatory sequences that could play a role in the pathobiology of fibroids. Although the vast majority of tumors including those with t(12;14) will have usual type histology, a study of a variant histology, plexiform uterine leiomyomata, has revealed dysregulation of HMGA2 through various rearrangements including the t(12;14).38
{kind=link}
The del(7)(q22q32) Subgroup
Del(7)(q22q32), an interstitial deletion of chromosome 7 involving bands q22-q32, is a common chromosome abnormality of uterine leiomyomas, with an observed frequency of approximately 17% in karyotypically abnormal fibroids.39, 40, 41 Loss of genetic material from 7q and rearrangements specifically involving band q22 have been found more consistently in uterine leiomyomas than in any other solid tumor. The finding of this particular anomaly as the sole alteration in some fibroids indicates its possible role as an early genetic event. Interestingly, del(7) can be observed simultaneously with t(12;14), suggesting involvement of del(7) in the karyotypic evolution of fibroids, although the t(12;14) can be similarly observed as an isolated aberration.42 Chromosome 7 rearrangements other than deletions have also been documented in fibroids and include various translocations that implicate 7q22 as the pathogenetic region.40
As is often the case in the identification of pathogenetic chromosomal segments, cytogenetic translocations involving chromosome 7 have been instrumental in narrowing the location of critical genes. Early studies reported deletions of chromosome 7 from band q11.23 to q36.43, 44, 45 With regard to mapping disease genes, this region, on the order of two megabases of DNA, represented an inordinately large distance in which to search. Further work limited the minimal region to band q22, and an additional study reporting three leiomyomas with translocations involving 7q22 confirmed the importance of this region to leiomyomas.39, 40, 46 Interestingly, one report found a subset of tumors with two discrete deletions mapping to the long arm of chromosome 7.47
It is notable that cultured fibroid specimens with chromosome 7 deletions or translocations are usually found in the mosaic state with 46,XX karyotypically normal cells.48 These cultures appear to be highly unstable and frequently lose the chromosomally aberrant line. Interestingly, cells with the del(7)(q22q32) appear to be more stable in culture when the t(12;14) is also present. The instability of del(7)(q22q32) cells without chromosome 12 aberrations is intriguing in light of the observation that fibroids with chromosome 12 abnormalities or rearrangements are often larger than tumors with chromosome 7 abnormalities, and larger fibroids are more likely to be chromosomally abnormal than smaller ones.15 Observations of tumor instability in culture and decreased tumor size in association with the del(7)(q22q32) suggest that a gene regulating cellular growth may reside in 7q22.
Interstitial deletions of the long arm of chromosome 7, as well as translocations involving the same region, have been reported in other benign mesenchymal tumors, including lipomas and endometrial polyps.29, 49, 50 In certain instances, the chromosome 7 abnormality has been the only anomaly present in the tumor. Interestingly, 7q deletions have also been documented in a subset of patients with primary acute nonlymphocytic leukemia (7.6%), with or without subsequent chemotherapy, and in those with myelodysplastic syndrome (19%). These specific deletions, considered a poor prognostic factor for certain hematological disorders/neoplasias, are observed with even greater frequency in patients with secondary acute myelocytic leukemia (26.8%) and secondary myelodysplastic syndrome (41%).43, 51, 52, 53 In one study, fluorescence in situ hybridization (FISH) analysis with markers in the 7q21-q31 region was used to study an established cell line from a leukemia patient and to limit the critical region in acute nonlymphocytic leukemia to a two-megabase interval.54 Patients with this particular cytogenetic abnormality are more likely to experience remissions of limited duration and have disease refractory to therapy. In sharp contrast, 7q deletions in benign mesenchymal tumors appear to have no adverse clinical significance.
Dissection of 7q22 to study leiomyoma-specific sequences is complicated by the fact that this is a gene-dense region including genes involved in developmental processes (DLX5, DLX6) and collagen metabolism (collagen type 1, procollagen C-endopeptidase enhancer), as well as those that encode acetylcholinesterase, plasminogen activator inhibitor type 1, and mucin.55, 56 A potential tumor-suppressor gene, CUTL1, which encodes a homeodomain protein, has been mapped to 7q22, and loss of heterozygosity (7 of 50 samples) or reduced expression (8 of 13 samples) has been demonstrated in a subset of leiomyomas.57 The DNA-replication initiation factor ORC5L has also been mapped to 7q22 and is deleted in a subset of leiomyomas.58 Because ORC5 protein is a subunit of the DNA replication complex so intimately connected to cell growth, it is tempting to speculate that loss of this protein may somehow contribute to instability and poor viability of leiomyomas with 7q22 deletions. Despite identification of several intriguing positional candidate genes, to date none has been proved to have a causative role in the genesis of leiomyomas. Clearly, because the region is both physically large and gene-rich, identification of pathogenetic sequences is a task of considerable magnitude. Most recently, molecular analysis of band 7q22 in uterine leiomyomas has defined a 10-centiMorgan critical region common to tumors with cytogenetic deletions.59 Taken together, these findings indicate an important role for a genetic locus at 7q22 in the development of uterine leiomyomas. Although the discovery of tumors in which the sole documented cytogenetic abnormality is del(7)(q22q32) may support an early causative role for this mutation, it is clear that additional submicroscopic mutations affecting other loci cannot yet be excluded.
Rearrangements of Chromosome 6 in Band p21
Rearrangements of band 6p21 have been observed frequently in the same group of previously mentioned mesenchymal tumors, including lipomas, pulmonary chondroid hamartomas, endometrial polyps, and uterine leiomyomas (see Fig. 1).60, 61, 62, 63 In fibroids, these rearrangements occur with a frequency of less than 5% and include t(6;14), t(6;10), and t(4;6), as well as inversions and translocations involving other chromosomal partners. After the architectural factor HMGA2 was identified as a gene involved in the pathobiology of this group of benign tumors, and given the observed rearrangements involving 6p21 in a subset of tumors, a high-mobility group (HMG) family member, HMGA1 [formerly designated as HMGIY], became an attractive positional candidate gene for fibroids. A single genomic clone including HMGA1 was found to span the breakpoint in a uterine leiomyoma with a complex rearrangement involving 6p21.3.64 Subsequent studies demonstrated rearrangement of the HMGA1 locus in hamartoma of the breast,62 a pericentric inversion of chromosome 6 involving band p21 in a uterine leiomyoma,61 and a translocation leading to an intergenic fusion with the LAMA4 gene (laminin α4) in a pulmonary chondroid hamartoma.60
OTHER CYTOGENETIC ABNORMALITIES
A variety of other cytogenetic abnormalities has been reported in leiomyomas, but with lower frequency. Among these are cases affecting the X chromosome, including del(X)(p11.2), t(X;12)(p22.3q15), -X, der(5)t(X;5)(p11;p15), del(X)(q12), der(X)t(X;3)(p22.3;q11.2), and inv(X)(p22q13).11, 44, 65, 66, 67, 68, 69 Although both p and q arms of the X chromosome are involved in these cases, it appears that the region of Xp11-p22 may be preferentially rearranged. No obvious relevant candidate genes have been identified in this region to date.
There have also been reports of structural rearrangements involving chromosomes 1 and 3 in leiomyomas, particularly in the form of ring chromosome 1, such as r(1)(p34q32). Ring chromosomes have been found in a few fibroid tumors, but they often occur concomitantly with other chromosomal changes and are therefore thought to represent secondary abnormalities.66, 70, 71 Other structural aberrations of chromosome 1 seen in leiomyomas include t(1;6)(q23;p21) and t(1;2)(p36;p24).72, 73 Additionally, heterogeneous rearrangements of both long and short arms of chromosome 1 have been described in uterine leiomyomata, with breakpoints distributed along both arms. The significance of these rearrangements of chromosome 1 to the pathogenesis of uterine leiomyomata is complicated by their frequent observation in other malignancies.74 However, loss of function mutations in the fumarate hydratase (FH) gene were found as the cause for autosomal dominantly inherited multiple cutaneous and uterine leiomyomatosis and hereditary leiomyomatosis and renal cell cancer, which previous linkage studies had mapped to 1q42.3-q43.75, 76, 77 The FH gene encodes an enzyme in the tricarboxylic acid (Krebs) cycle.78 FH acts as a tumor suppressor and its measured activity is very low to absent in tumors from individuals with syndromic leiomyomatosis.76 Evaluation of the role of FH in sporadic leiomyomas and leiomyosarcomas suggests that mutations in fumarate hydratase are rare in these tumors and therefore do not play a major role in the development of leiomyosarcomas or nonsyndromic uterine leiomyomas,79but no doubt underlie some disease as evinced also by genetic linkage analysis.80 To date, no other candidate genes on chromosome 1 have been identified as having a role in leiomyoma formation. From a subset of more than 800 karyotyped uterine leiomyomas, a group of nine cases exhibiting near-diploid karyotypes with loss of almost the entire short (p) arm of chromosome 1 [i.e. del(1)(p11p36)] was observed often associated with other aberrations, particularly loss of chromosomes 19 and/or 22.81 Of eight tumors for which the histologic diagnosis was known, four were diagnosed as cellular and transcriptional profiles of tumors with 1p deletion were more similar to those of leiomyosarcoma than to profiles of myometrium and uterine leiomyoma, as determined by hierarchical cluster analysis. The similarity between the transcriptional profiles suggests the possibility of a common pathogenetic mechanism. A number of rearrangements of chromosome 3 have been found in leiomyomas, both as sole abnormalities and as those accompanying other rearrangements, including ins(2;3)(q31;p12p25), del(3)(p14), del(3)(q24), and t(3;7)(p11;p11).67
Rearrangement of 10q22 is a recurring chromosomal aberration in uterine leiomyoma, and chromosome 10 breakpoints have been mapped within the third intron of MORF (monocytic leukemia zinc finger protein-related factor or MYST4) in four uterine leiomyomata.82 MORF is a member of the MYST family of histone acetyltransferase and previously has been found rearranged in some types of acute myeloid leukemia. Monosomy 10 and deletions affecting the long arm (especially band q22) have been observed in fibroids. Two tumor suppressor genes are known to map to this area. Both the PTEN/MMAC1 gene (10q23.3) and the DMBT1 gene (10q25.3–26.1) are located on the long arm of chromosome 10, but neither has been shown to contribute to the pathogenesis of fibroid tumors.83, 84 Finally, trisomy 12 has also been reported in leiomyomas and may act to increase the dosage of genes such as HMGA2, as described in the next section.12
INVOLVEMENT OF THE HMGA FAMILY OF GENES IN LEIOMYOMAS
The architectural factor gene HMGA2 (at 12q15) was initially considered a positional candidate gene for leiomyomas after a series of FISH experiments identified a YAC that mapped to 12q15 and spanned the translocation breakpoints in uterine fibroid, pulmonary chondroid hamartoma, and lipoma specimens. HMGA2 had been mapped earlier to mouse chromosome 10, a segment of which is homologous to human 12q13–15.85 The relevance of these mapping data became apparent when transgenic mouse mutants were generated and showed aberrant growth leading to a pygmy phenotype, caused by disruption of the murine HMGA2 locus.86 These data, along with experiments showing a requirement for HMGA2 in cellular transformation, and the understanding that the protein acts as an accessory transcriptional factor, combined to make HMGA2 an intriguing candidate gene in the study of mesenchymal tumors with t(12;14) rearrangements.87, 88 In a study of lipoma specimens with 12q15 rearrangements, FISH analysis and rapid amplification of cDNA ends (RACE) experiments demonstrated that HMGA2 was in fact disrupted by the translocation and that chimeric fusion transcripts were produced.35
Soon afterward, HMGA1 (at chromosome 6p21, another site of consistent rearrangement in benign mesenchymal tumors) was evaluated as a candidate gene, and determined to be involved in these tumors. Along with HMGA2, the HMGA1 protein belongs to the heterogeneous HMG family of nonhistone DNA-binding proteins. Within this family, three categories have been recognized: the HMG box containing HMGB-1/HMGB-2 (formerly HMG-1 and formerly HMG-2, respectively) proteins; the active chromatin-associated HMGN1/HMGN2 (formerly HMG-14 and formerly HMG-17, respectively) proteins; and the HMGA (formerly HMGI) proteins HMGA2 and HMGA1.89 As a group, HMGA proteins are known to bind to the minor groove of AT-rich DNA through a domain known as the AT hook. Through a combination of protein–protein and protein–DNA interactions, HMGA proteins appear to affect gene activity by modulating DNA conformation and regulating access of transcription factors to target genes. Both HMGA2 and HMGA1 have been shown to be phosphorylated by p34/cdc2-like kinases in a cell cycle-dependent manner, subsequently modulating the affinity of HMGA2 protein for DNA.90, 91 Of the HMGA proteins, HMGA1 is the best characterized and has been shown to stimulate binding of NF-kB and ATF2 to the interferon B promoter by changing DNA conformation. As a result of HMGA1 action, NF-kB and ATF2 are able to interact with each other and with the basal transcriptional machinery. AT-rich sequences in the promoters of E-selectin, interleukin-2 receptor α-chain, the chemokine MGSA/GRO, and the class II major histocompatibility complex gene HLA-DRA have all been demonstrated to bind specifically and directly to the HMGA1 protein.
{kind=link}
HMGA2 and HMGA1 are both closely related as well as highly conserved (96% conservation between mouse and human HMGA2 homologues).92, 93 Sequence analysis has shown that the first three HMGA2 exons are clustered within 13 kilobases of each other and together encode a DNA-binding domain comprising the AT hook motif (Fig. 2). A large intron (approximately 140 kilobases) between exons 3 and 4 separates the DNA-binding domains from the rest of the protein. Exon 4 encodes an 11-amino acid spacer domain that is absent from HMGA1 and precedes a 13-kilobase intron that separates it from the acidic domain encoded by exon 5. The entire HMGA2 gene spans approximately 160 kilobases, including a 327-base pair coding region, an 854-base pair 5' untranslated region (UTR) and a 2966-base pair 3' UTR, which together encode a 4.1-kilobase mRNA. In contrast, the smaller (10 kilobase) HMGA1 gene consists of eight exons: exons 1 to 4 are noncoding, exons 5 to 7 contain the AT hooks corresponding to exons 1 to 3 of HMGA2, and exon 8 encodes the acidic C-terminus of the protein. Alternate splicing of this gene results in two products, one of which is a variant lacking a 33-basepair segment between exons 5 and 6 and which is also absent from HMGA2. Despite the great structural similarity between HMGA2 and HMGA1, expression patterns of the two proteins are strikingly dissimilar, perhaps suggesting the existence of distinct regulatory elements as well as different functional roles. In particular, it has been noted that increased levels of HMGA1 correlate with a malignant phenotype in a variety of tumors, including those of murine lung, thymus, and mammary epithelial cells.94 In human neoplasms, increased HMGA1 expression has been demonstrated in high-grade prostate tumors, thyroid carcinoma, and colorectal carcinomas. However, HMGA1 expression has also been seen in nonneoplastic animal cells, including rat fibroblasts and thyroid epithelial cells, as well as in numerous nonneoplastic human tissues, including heart, brain, lung, skeletal muscle, kidney, pancreas, spleen, thymus, prostate, testis, ovary, small intestine, colon, and leukocytes.92, 95
In contrast, HMGA2 shows a limited expression pattern in a multitude of tissues and organs during murine embryogenesis with the notable exception of the brain, in which HMGA2 is expressed in a very restricted portion of the forebrain.96 In additional studies, RT-PCR analysis revealed expression of HMGA2 in many fetal tissues, including adrenal, aorta, bone, brain, heart, intestine, kidney, liver, lung, muscle, ovary, placenta, skin, spleen, stomach, testis, and uterus. In contrast, HMGA2 expression in adult tissue was detected only in lung and kidney.97 It has been previously reported that HMGA2 expression is not detected in myometrium or karyotypically normal uterine leiomyomata. However, uterine leiomyomas with chromosomal rearrangements of 12q15 frequently express HMGA2. Recent data have demonstrated that HMGA2 expression was detectable in karyotypically normal uterine leiomyomas as well as in matched myometrium.98 The level of HMGA2 expression in karyotypically normal uterine leiomyomas was similar to the level of expression in myometrium; however, it was significantly less than the level measured in uterine leiomyomas with 12q15 rearrangements. These results refine the prevailing thought that HMGA2 is not detectable in myometrium and karyotypically normal uterine leiomyomas. Furthermore, these findings suggest that HMGA2 expression is required in normal adult myometrial physiology. A number of studies have shown a relationship between elevated HMGA2 expression and cellular transformation. In rat fibroblasts, HMGA2 expression can be induced after transformation with viral oncogenes, suggesting a role in maintenance of the transformed state.99, 100, 101 Indeed, support for this notion comes from in vitro studies demonstrating inhibition of cellular transformation and reversal of the tumorigenic phenotype after suppression of HMGA2 expression.88 Such data are consistent with a role for HMGA2 in regulation of cellular proliferation.
Role of HMGA2 and HMGA1 Fusion Transcripts
Involvement of both HMGA2 and HMGA1 in molecular cytogenetic rearrangements and production of ectopic fusion transcripts has been demonstrated in a variety of mesenchymally derived tumors. In particular, chimeric transcripts identified in lipoma specimens include one consisting of the amino terminus of HMGA2 fused to transcriptional regulatory domains (corresponding to the LIM proteins) at the carboxy end of the protein.35, 102 A number of LIM family proteins act as transcription factors whose activity is modulated by protein–protein dimers formed through LIM domains. Any consideration of the biology of these novel fusion proteins must include the possibility that HMGA2 is endowed with novel transcriptional regulatory abilities in addition to the DNA-binding ability already provided by the AT hook domains. Clearly, dysregulated HMGA proteins contribute to the pathobiology of these benign mesenchymal tumors.
In many cases, HMGA2 is interrupted at the end of the third exon within the large 140-kilobase intron separating the AT hook domains from the fourth exon. Fusion transcripts of this sort have been demonstrated in lipomas, uterine leiomyomas, and pulmonary chondroid hamartomas.
In lipomas, one member of the LIM protein gene family, LPP (lipoma preferred partner), has been observed frequently in the formation of HMGA2/LPP fusion transcripts after a t(3;12) rearrangement.89, 103 Of 23 lipomas tested, eight expressed this fusion transcript consisting of the DNA-binding domains of HMGA2 fused with the protein dimerization domains of LPP. An inability to detect the reciprocal fusion products suggests a role for the HMGA2/LPP fusion product in lipoma development.
In contrast to the events observed in lipomas, a significant number of leiomyomas with cytogenetic aberrations affecting the HMGA2 locus show breakpoints outside the coding region of the gene. In a series of seven leiomyomas, genomic breaks were found to occur from 10 kilobases to greater than 100 kilobases upstream of HMGA2.104 Such extragenic breaks suggest the disruption of regulatory sequences, which leads to abnormal expression of HMGA2. This is not an unprecedented mechanism for gene disruption, because breakpoints up to 100 kilobases upstream of the MYC gene have been shown to alter expression and lead to neoplasia in Burkitt lymphoma.105 A similar phenomenon occurs at the BCL1 locus (near the CCND1 gene) in B-cell and mantle-cell lymphomas. In this instance, BCL1 is disrupted through cytogenetic rearrangements, primarily translocations, involving chromosome 11, band q13. As a result of these translocations, cyclin D1 (CCND1) expression is activated ectopically, and consequently promotes tumorigenesis.106, 107
A previous study of a pleomorphic salivary gland adenoma with a complex chromosomal rearrangement involving 12q15 showed a break in the 3'UTR of HMGA2.108 The 3'UTR of HMGA2 normally contains eight AUUUA sequence motifs, which are thought to play a role in the destabilization and degradation of mRNA. The fusion transcript of the salivary gland adenoma showed preservation of the entire HMGA2 coding region, but the mRNA-destabilizing AUUUA motifs were replaced with a novel sequence. A scenario may be envisioned in which an increasingly stable HMGA2 mRNA leads to elevated levels of protein, thereby contributing to dysregulation of cellular growth. Novel sequences have been reported to be fused to HMGA2 in five lipomas, a pulmonary chondroid hamartoma, and an angiomyxoma, all of which showed a pericentric inversion of chromosome 12, inv(12)(p11.2q15). The nature and function of this sequence on 12p, as well as its contribution to tumorigenesis, are unknown.109
Although several known genes have been shown to be involved as partners in these rearrangements (e.g. a mitochondrial alcohol dehydrogenase–HMGA2 fusion in a single fibroid;110 an RTVL-H 3' LTR sequence fused with HMGA2 in a fibroid and two cases of pulmonary chondroid hamartoma; and a LAMA4–HMGA1 fusion in another case of pulmonary chondroid hamartoma),60 most fusion partners have involved uncharacterized, novel DNA sequences.35, 102 In fact, for HMGA2, most chimeric transcripts result from the addition of a few amino acids (i.e. 1 through 10) to exon 3. Consequently, one possibility that must be considered is that truncation of HMGA2 mRNA, instead of fusion to ectopic sequences, is a primary pathogenetic event in the development of mesenchymal neoplasms in these tumors. Another fusion partner of HMGA2 in fibroids is RAD51L1.36, 37 Localization of the chromosome 12 breakpoints relative to the HMGA2 locus in a panel of uterine leiomyomas with 12q15 rearrangements demonstrated that most rearrangements mapped in the 5' region of HMGA2. HMGA2–RAD51L1 transcripts were detected in four tumors: two of these had uncommon rearrangements in the 3' UTR region of HMGA2, and the other two had 5' rearrangements. The fusion transcripts detected contained the full coding sequence of HMGA2.111 The mechanism of fusion transcripts derived from tumors with 5' breakpoints was unclear; however, mosaicism for both 5' and 3' rearrangements was not ruled out. The infrequency of fusion transcripts as well as the fact that the fusion transcripts seen contained the full coding sequence of HMGA2 suggests that formation of a fusion transcript is not the principal pathobiological mechanism in uterine fibroids and that dysregulated expression of HMGA2 may be more important for tumor growth. Nonetheless, because HMGA2–RAD51L1 fusions were not observed in all cases, and because HMGA2 transcripts were present in all tumors with t(12;14), it remains to be determined whether the HMGA2–RAD51L1 transcript is a critical biological event for uterine leiomyomas or whether dysregulated HMGA2 expression itself is the important molecular mechanism. Given that a significant proportion of breaks in leiomyomas actually occur either 5' (10–100 kilobases upstream) or, in a few cases, 3' of HMGA2, this further supports that dysregulation of HMGA2 expression may be an important mechanism of tumor growth. It remains to be determined whether creation of fusion mRNAs, truncation of HMGA2, or disruption of regulatory sequences constitutes the major pathogenetic event in the genesis of benign mesenchymal tumors.
What primary role are HMGA proteins fulfilling in benign tumors? Given their function as architectural factors, which remodel regions of the double helix, one strong possibility is that they provide genomic stability. In this capacity, HMGA proteins may be responsible for regulating access to target genes of requisite transcription factors. Whether the binding of transcription factors required for the expression of growth-promoting genes is facilitated, or whether the binding of factors necessary for the activation of tumor suppressor genes is prevented, the growth and development of a tumor may be enhanced.
GENETIC SUSCEPTIBILITY FOR LEIOMYOMAS
Evidence for a genetic predisposition to uterine leiomyoma development comes from epidemiological studies including familial aggregation,112 racial prevalence,113, 114 and twin studies,115 as well as from genetic linkage studies in families with uterine leiomyomata-associated heritable syndromes.75, 76 Literature describing familial clustering of uterine leiomyomas first appeared in 1938, when a German group reported that uterine leiomyomas were 4.2 times more frequent among first-degree relatives of affected probands than among first-degree relatives of unaffected probands.112 In the late 1980s, a series of Russian studies provided further evidence for familial aggregation of uterine leiomyomas: sisters, daughters, and mothers of affected probands had a 2.3-, 2.0, and 1.6-fold increased risk for uterine leiomyomas, respectively, over the general population.116, 117, 118 Of note, maternal data were determined by the authors to be unreliable, likely accounting for the lower risk of 1.6. These results were confirmed by a 1995 Russian study, which showed that uterine leiomyomas were 2.2 times more frequent among first-degree relatives in families with two or more verified fibroid cases.116, 117, 118 Schwartz and coauthors found that the odds ratio for uterine leiomyomas among first-degree relatives of affected probands, compared with relatives of unaffected probands, was 2.5.119 This odds ratio increased to 5.7 after stratifying cases by age of proband (younger than age 45 years) and of relatives (younger than age 40 years) (Fig. 3).
{kind=link}
Further evidence for a genetic susceptibility to uterine leiomyomas comes from several racial predisposition studies showing that black women experience a three- to nine-fold higher incidence of uterine leiomyomas than white women.113, 114 Racial differences in known risk factors do not explain the higher rate of uterine leiomyomas in black women. Moreover, black women experience an earlier age of fibroid diagnosis, a higher hysterectomy rate for uterine leiomyomas, larger and more numerous uterine leiomyomas, and more severe symptoms than white women.10
Additional evidence for the heritability of uterine leiomyomas can be inferred from hysterectomy data in the Australian twin registry. A 1992 analysis of this data revealed that the twin-pair correlation for hysterectomy in monozygous twins (r = 0.65 ± 0.05) was twice that of dizygous twins (r = 0.32 ± 0.09), wholly consistent with the expected rates for a genetically influenced trait.115 Although conditions other than uterine leiomyomas may contribute to hysterectomy, this finding in twins strongly suggests a genetic liability for uterine leiomyomas, because these tumors are the most common indication for hysterectomy. These data were supported by a Finnish twin cohort study, which found that case-wise concordance for being hospitalized for uterine leiomyomas was higher in monozygous twins (0.31; 95% confidence interval [CI]: 0.24–0.37) than dizygous twins (0.18; 95% CI: 0.14–0.22).120 Overall, heritability for uterine leiomyomas has been estimated to be 0.26, 0.69, and 0.79 for populations in Finland, United Kingdom, and Russia, respectively.117, 120, 121
Finally, there are several syndromes of genetic interest associated with uterine leiomyomas. Two such inherited disorders are Reed syndrome (OMIM 150800), characterized by uterine leiomyomas in association with multiple cutaneous leiomyomata, and hereditary leiomyomatosis and renal cell cancer (HLRCC) (OMIM 605839), a cancer syndrome characterized by uterine leiomyomas and papillary renal cell carcinoma.122, 123, 124 Two independent studies performed on 11 families with Reed syndrome and two families with HLRCC identified a putative susceptibility gene on chromosome 1 in bands q42-44,75 and the gene was identified as FH. Whole-gene deletions to point mutations were detected, and assays of FH revealed decreased activity in heterozygotes. The role of a gene involved in intermediary metabolism is somewhat surprising, although not without precedent, because succinate dehydrogenase has been found to be pathogenetic in some paragangliomas.125
Taken together, these data strongly suggest a genetic component in the etiology of fibroids. Clearly, an important avenue of research involves studying affected women and their first-degree relatives who also have uterine fibroids. The availability and examination of such persons ultimately not only will hasten cytogenetic and molecular studies but also will be crucial to dissecting and defining the genetic loci that undoubtedly contribute to the development of uterine leiomyomas.
REFERENCES
National Center for Health Statistics: Hysterectomies in the United States. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control1987 |
|
Cramer SF, Patel A: The frequency of uterine leiomyomas. Am J Clin Pathol 94:435-438, 1990 |
|
Candiani GB, et al: Risk of recurrence after myomectomy. Br J Obstet Gynaecol 98:385-389, 1990 |
|
Cotran RS, Kumar V, Robbins SL: Robbins Pathologic Basis of Disease. Philadelphia, WB Saunders, 1999 |
|
Brandon DD, Strawn EY, et al: Progesterone receptor messenger ribonucleic acid and protein are overexpressed in human uterine leiomyomas. Am J Obstet Gynecol 169:(78):1993 |
|
Rein MS, Barbieri RL, Friedman AJ: Progesterone: A critical role in the pathogenesis of uterine myomas. Am J Obstet Gynecol 172:(1):14-18, 1995 |
|
Garcia Muret MP, et al: Familial leiomyomatosis cutis et uteri (Reed's syndrome). Arch Dermatol Res 280:S29-S32, 1988 |
|
Meilahn EN, et al: Characteristics of women with hysterectomy. Maturitas 11:319-329, 1989 |
|
Kjerulff KH, et al: Hysterectomy and race. Obstet Gynecol 82:757-764, 1993 |
|
Huyck KL, Panhuysen CI, Cuenco KT, et al. The impact of race as a risk factor for symptom severity and age at diagnosis of uterine leiomyomata among affected sisters. Am J Obstet Gynecol 2008;198(2):168 e1-9. |
|
Nilbert M, Heim S: Uterine leiomyoma cytogenetics. Genes Chromosomes Cancer 2:3-13, 1990 |
|
Rein MS, et al: Cytogenetic abnormalities in uterine leiomyomata. Obstet Gynecol 77:923-926, 1991 |
|
Brosens I, et al: Clinical significance of cytogenetic abnormalities in uterine myomas. Fertil Steril 69:232-235, 1998 |
|
Pandis N, et al: Histologic-cytogenetic correlations in uterine leiomyomas. Int J Gynecol Cancer 1:163-168, 1991 |
|
Rein MS, et al: Cytogenetic abnormalities in uterine myomas are associated with myoma size. Mol Hum Reprod 4:83-86, 1998 |
|
Mashal RD, et al: Analysis of androgen receptor DNA reveals the independent clonal origins of uterine leiomyomata and the secondary nature of cytogenetic aberrations in the development of leiomyomata. Genes Chromosomes Cancer 11:1-6, 1994 |
|
Hashimoto K, Kamiura S et. al: Clonal determination of uterine leiomyomas by analyzing differential inactivation of the X-chromosome-linked phosphoglycerokinase gene. Gynecol Obstet Invest 40:(204):1995 |
|
Linder D: Glucose-6-phosphate dehydrogenase mosaicism: Utilization as a cell marker in the study of leiomyomas. Science 150:(67):1965 |
|
Townsend DE, et al: Unicellular histogenesis of uterine leiomyomas as determined by electrophoresis of glucose-6-phosphate dehydrogenase. Am J Obstet Gynecol 107:1168-1173, 1970 |
|
Nilbert M: Independent origin of uterine leiomyomas with karyotypically identical alterations. Gynecol Obstet Invest 33:(246):1992 |
|
Ozisik YY, Powell M, et al: Chromosome 7 biclonality in uterine leiomyoma. Cancer Genet Cytogenet 67:(59):1993 |
|
Cavenee WK, James CD: Molecular genetics of human cancer predisposition and progression. Mutation Res 247:(199):1991 |
|
Goyette MC, Fasching CL, et al: Progression of colorectal cancer is associated with multiple tumor suppressor gene defects but inhibition of tumorigenicity is accomplished by correction of any single defect via chromosome transfer. Mol Cell Biol 12:(1387):1992 |
|
Meloni AM, et al: Uterine leiomyomas: Cytogenetic and histologic profile. Obstet Gynecol 80:209-217, 1992 |
|
Kazmierczak B, et al: Cytogenetic and molecular analysis of an aggressive angiomyxoma. Am J Pathol 147:580-585, 1995 |
|
Calabrese G, et al: Chromosome abnormalities in breast fibroadenomas. Genes Chromosomes Cancer 3:202-204, 1991 |
|
Ozisik YY, et al: Chromosome abnormalities in breast fibroadenomas. Cancer Genet Cytogenet 77:125-128, 1994 |
|
Vanni R, et al: Endometrial polyp: Another benign tumor characterized by 12q13-q15 changes. Cancer Genet Cytogenet 68:32-33, 1993 |
|
Dal Cin P, et al: Four cytogenetic subgroups can be identified in endometrial polyps. Cancer Res 55:1565-1568, 1995 |
|
Mandahl N, et al: Aberrations of chromosome segment 12q13-15 characterize a subgroup of hemangiopericytomas. Cancer 71:3009-3013, 1993 |
|
Turc-Carel C, et al: Cytogenetic studies of adipose tissue tumors. I. A benign lipoma with reciprocal translocation t(3;12)(q28;q14) Cancer Genet Cytogenet 23:283-289, 1986 |
|
Fletcher JA, et al: Cytogenetic and histologic findings in 17 pulmonary chondroid hamartomas: Evidence for a pathogenic relationship with lipomas and leiomyomas. Genes Chromosomes Cancer 12:220-223, 1995 |
|
Bullerdiek J, et al: Rearrangements of chromosome region 12q13-15 in pleomorphic adenomas of the human salivary gland (PSA). Cytogenet Cell Genet 45:187-190, 1987 |
|
Schoenberg Fejzo M, et al: Identification of a YAC spanning the translocation breakpoints in uterine leiomyomata, pulmonary chondroid hamartoma and lipoma: physical mapping of the 12q14-q15 breakpoint region in uterine leiomyomata. Genomics 26:265-271, 1995 |
|
Ashar HR, et al: Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell 82:57-65, 1995 |
|
Ingraham SE, et al: hREC2, a RAD51-like gene, is disrupted by t(12;14)(q15;q24.1) in a uterine leiomyoma Cancer Genet Cytogenet 115:56-61, 1999 |
|
Schoenmakers EFPM, Huysmans C, Van de Ven WJM: Allelic knockout of novel splice variants of human recombination repair gene RAD51B in t(12;14) in uterine leiomyomas. Cancer Res 59:19-23, 1999 |
|
Hodge JC, Quade BJ, Rubin MA, Stewart EA, Dal Cin P, Morton CC. Molecular and cytogenetic characterization of plexiform leiomyomata provide further evidence for genetic heterogeneity underlying uterine fibroids. Am J Pathol 2008;172(5):1403-10. |
|
Ozisik YY, et al: Deletion 7q22 in uterine leiomyoma. A cytogenetic review Cancer Genet Cytogenet 71:1-6, 1993 |
|
Sargent MS, et al: Translocations in 7q22 define a critical region in uterine leiomyomata. Cancer Genet Cytogenet 77:65-68, 1994 |
|
Ishwad CS, et al: Molecular and cytogenetic analysis of chromosome 7 in uterine leiomyomas. Genes Chromosomes Cancer 14:51-55, 1995 |
|
Sait SN, Ovanessoff S, Sandberg AA: A uterine leiomyoma showing both t(12;14) and del(7) abnormalities. Cancer Genet Cytogenet 37:(157):1989 |
|
Pandis N, et al: Parallel karyotypic evolution and tumor progression in uterine leiomyoma. Genes Chromosomes Cancer 2:311-317, 1990 |
|
Fan SX, Berger CS, et al: Cytogenetic findings in nine leiomyomas of the uterus. Cancer Genet Cytogenet 47:(179):1990 |
|
Vanni R., Lecca U, Faa G: Uterine leiomyoma cytogenetics II: Report of forty cases. Cancer Genet Cytogenet 53:247-256, 1991 |
|
Ozisik YY, et al: Chromosome 7 biclonality in uterine leiomyoma. Cancer Genet Cytogenet 67:59-64, 1993 |
|
Ishwad CS, et al: Two discrete regions of deletion at 7q in uterine leiomyomas. Genes Chromosomes Cancer 19:156-169, 1997 |
|
Xing YP, Powell WL, Morton CC: The del(7q) subgroup in uterine leiomyomata: Genetic and biologic characteristics. Cancer Genet Cytogenet 98:69-74, 1997 |
|
Sreekantaiah C, Sandberg AA: Clustering of aberrations to specific chromosome regions in benign neoplasms. Int J Cancer 48:194-198, 1991 |
|
Dal Cin P, et al: Deletion of the long arm of chromosome 7 in lipoma. Cancer Genet Cytogenet 96:85-86, 1997 |
|
Pandis N, et al: Chromosome analysis of 96 uterine leiomyomas. Cancer Genet Cytogenet 55:11-18, 1991 |
|
Litt M, Sharma V: Shadow bands seen when typing polymorphic dinucleotide repeats: Some causes and cures. Biotechniques 15:(280):1993 |
|
Davidovitz Y, et al: Deletion of the long arm of chromosome 7 in myelomonocytic leukemia with bone marrow eosinophilia. Cancer Genet Cytogenet 97:122-124, 1997 |
|
Tosi S, Rambaldi A, et al: Characterization of 7q abnormalities in the human myeloid leukemia cell line GFD8 by fluorescence in situ hybridization (FISH). Cytogenet Cell Genet 71:(28):1995 |
|
Vachon G, et al: Homeotic genes of the Bithorax complex repress limb development in the abdomen of the Drosophila embryo through the target gene Distal-less. Cell 71(3):437-450, 1992 |
|
Tsui LC, Donis-Keller H, Grzeschik KH: Report of the second international workshop on human chromosome 7 mapping 1994. Cytogenet Cell Genet 71:(1):2-21, 1995 |
|
Zeng WR, et al: Loss of heterozygosity and reduced expression of the CUTL1 gene in uterine leiomyomas. Oncogene 14:2355-2365, 1997 |
|
Ishiai M, et al: Isolation of human and fission yeast homologues of the budding yeast origin recognition complex subunit ORC5: Human homologue (ORC5L) maps to 7q22. Genomics 46:294-298, 1997 |
|
Sell SM, et al: Molecular analysis of chromosome 7q21.3 in uterine leiomyoma: analysis using markers with linkage to insulin resistance Cancer Genet Cytogenet 100:165-168, 1998 |
|
Xiao S, et al: HMGI(Y) activation by chromosome 6p21 rearrangements in multilineage mesenchymal cells from pulmonary hamartoma. Am J Pathol 150:901-910, 1997 |
|
Williams AJ, et al: HMGI(Y) expression in human uterine leiomyomata: Involvement of another HMG architectural factor in a benign neoplasm. Am J Pathol 150:911-918, 1997 |
|
Dal Cin P, Christiaens MR, et al: Hamartoma of the breast with involvement of 6p21 and rearrangement of HMGI(Y). Cancer 20:(90):1997 |
|
Tallini G, et al: Expression of HMGI-C and HMGI(Y) in ordinary lipoma and atypical lipomatous tumors. Am J Pathol 151:37-43, 1997 |
|
Kazmierczak B, et al: PAC clone containing the HMGI(Y) gene spans the breakpoint of a 6p21 translocation in a uterine leiomyoma cell line. Genes Chromosomes Cancer 17:191-193, 1996 |
|
Turc-Carel C, et al: Consistent breakpoints in region 14q22-q24 in uterine leiomyoma. Cancer Genet Cytogenet 32:25-31, 1988 |
|
Vanni R, Paoli R, Lecca U: Uterine leiomyoma cytogenetics: I. Rearrangements of chromosome 12 Cancer Genet Cytogenet 37:(49):1989 |
|
Nilbert M, et al: Characteristic chromosome abnormalities, including rearrangements of 6p, del(7q), +12, and t(12;14), in 44 uterine leiomyomas. Hum Genet 85:605-611, 1990 |
|
Mark J, et al: Chromosomal patterns in human benign uterine leiomyomas. Cancer Genet Cytogenet 44:1-13, 1990 |
|
Ozisik YY, Surti MA, et al: Inversion (X)(p22q13) in a uterine leiomyoma. Cancer Genet Cytogenet 61:(131):1992 |
|
Nilbert M, Mandahl N, et al: Ring formation and structural rearrangements of chromosome 1 as secondary changes in uterine leiomyomas with t(12;14)(q14-15;q23-24). Cancer Genet Cytogenet 36:(183):1988 |
|
Casartelli C, Rogatto SR, Philbert PMP: A cytogenetic study of uterine leiomyomas. Cancer Genet Cytogenet 41:(279):1989 |
|
Havel G, Dahlenfors R, Mark J: Cytogenetic relationship between uterine lipoleiomyomas and typical leiomyomas. Virchows Arch A Pathol Anat Histopathol 57:(77):1989 |
|
Mark J, et al: Cytogenetical observations in human benign uterine leiomyomas. Anticancer Res 8:621-626, 1988 |
|
Dal Cin P, Morton CC: 1q42∼q44 is rarely cytogenetically involved in sporadic uterine leiomyomata. Cancer Genet Cytogenet 138:92-93, 2002 |
|
Alam NA, et al: Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q42.3-q43 Am J Hum Genet 68:(5):1264-1269, 2001 |
|
Tomlinson IP, et al: Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30:(4):406-410, 2002 |
|
Toro JR, Wei Ming-Hui NM, Warren MB, et al: Mutations in the Fumarate Hydratase Gene Cause Hereditary Leiomyomatosis and Renal Cell Cancer in Families in North America. Am J Hum Genet 73:2003 |
|
Kiuru M., Lehtonen R, Arola J, et al: Few FH Mutations in Sporadic Counterparts of Tumor Types Observed in Hereditary Leiomyomatosis and Renal Cell Cancer Families. Cancer Res 62:4554-4557, 2002 |
|
Barker K, Bevan S, Wang R, et al: Low frequency of somatic mutations in the FH/multiple cutaneous leiomyomatosis gene in sporadic leiomyosarcomas and uterine leiomyomas. Br J Cancer 87:(4):446-448, 2002 |
|
Gross KL, Panhuysen CI, Kleinman MS, et al. Involvement of fumarate hydratase in nonsyndromic uterine leiomyomas: genetic linkage analysis and FISH studies. Genes Chromosomes Cancer 2004;41(3):183-90. |
|
Christacos NC, Quade BJ, Cin PD, Morton CC. Uterine leiomyomata with deletions of Ip represent a distinct cytogenetic subgroup associated with unusual histologic features. Genes Chromosomes Cancer 2006;45(3):304-12. |
|
Moore SD, Herrick SR, Ince TA, et al. Uterine leiomyomata with t(10;17) disrupt the histone acetyltransferase MORF. Cancer Res 2004;64(16):5570-7. |
|
Steck PA, Jasser SA, et al: Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers Nat Genet 15:(356):1997 |
|
Mollenhauer J, Scheurlen W, et al: DMBT1, a new member of the SRCR superfamily, on chromosome 10q25.3-26.1 is deleted in malignant brain tumours Nat Genet 17:(32):1997 |
|
Justice MJ, et al: A genetic linkage map of mouse chromosome 10: Localization of eighteen molecular markers using a single interspecific backcross. Genetics 125:855-866, 1990 |
|
Zhou X, et al: Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 376:771-774, 1995 |
|
Manfioletti G, et al: cDNA cloning of the HMGI-C phosphoprotein, a nuclear protein associated with neoplastic and undifferentiated phenotypes. Nucleic Acids Res 19:6793-6797, 1991 |
|
Berlingieri MT, et al: Inhibition of HMGI-C protein synthesis suppresses retrovirally induced neoplastic transformation of rat thyroid cells. Mol Cell Biol 15:1545-1553, 1995 |
|
Hess JL: Chromosomal translocations in benign tumors: The HMGI proteins. Am J Clin Pathol 109:251-261, 1998 |
|
Reeves R, Langan TA, Nissen MS: Phosphorylation of the DNA-binding domain of nonhistone high-mobility group I protein by cdc2 kinase: Reduction of binding affinity. Proc Natl Acad Sci 88:1671-1675, 1991 |
|
Nissen MS, Reeves R: Phosphorylation by cdc2 kinase modulates DNA binding activity of high mobility group nonhistone chromatin protein. J Biol Chem 266:(19945):1991 |
|
Friedman M, et al: Organization, inducible-expression and chromosome localization of the human HMGI(Y) non-histone protein gene. Nucleic Acids Res 21:4259-4267, 1993 |
|
Ashar HR, et al: Genomic characterization of human HMGIC, a member of the accessory transcription factor family found at translocation breakpoints in lipomas. Genomics 31:207-214, 1996 |
|
Ram TG, Reeves R, Hosick HL: Elevated high mobility group-I(Y) gene expression is associated with progressive transformation of mouse mammary epithelial cells. Cancer Res 53:2655-2660, 1993 |
|
Tamimi Y, et al: Increased expression of high mobility group protein I(Y) in high grade prostatic cancer determined by in situ hybridization. Cancer Res 53:5512-5516, 1993 |
|
Rogalla P, et al: HMGI-C expression patterns in human tissues. Am J Pathol 149:775-779, 1996 |
|
Gattas GJF, et al: HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes Chromosomes Cancer 25:316-322, 1999 |
|
Gross KL, Manchanda N, Weremowicz S, et al: HMGA2 Expression in Uterine Leiomyomata and Myometrium: Quantitative Analysis and Tissue Culture Studies. Genes Chromosomes Cancer In Press 2003 |
|
Giancotti V, DiFiore PP, et al: Changes in nuclear proteins on transformation of rat epithelial thyroid cells by a murine sarcoma retrovirus. Cancer Res 45:6051, 1985 |
|
Giancotti V, et al: Elevated levels of a specific class of nuclear phosphoroproteins in cells transformed with v-ras and v-mos oncogenes and by co-transfection with c-myc and polyoma middle T genes. Eur Mol Biol Org J 6:1981-1987, 1987 |
|
Giancotti V, et al: Analysis of the HMGI nuclear proteins in mouse neoplastic cells induced by different procedures. Exp Cell Res 184:538-545, 1989 |
|
Schoenmakers EFPM, et al: Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat Genet 10:436-443, 1995 |
|
Petit MMR, et al: LPP, the preferred fusion partner gene of HMGIC in lipomas, is a novel member of the LIM protein gene family. Genomics 36:118-129, 1996 |
|
Schoenberg Fejzo M, et al: Translocation breakpoints upstream of the HMGIC gene in uterine leiomyomata suggest dysregulation of this gene by a mechanism different from that in lipomas. Genes Chromosomes Cancer 17:1-6, 1996 |
|
Holt JT, et al: Molecular mechanisms of hematological neoplasms. In: Stamatoyannopoulos G, et al (eds): The Molecular Basis of Blood Disease. pp 360-362, Philadelphia, WB Saunders, 1987 |
|
Lammie GA, Silver JB, et al: Proviral insertions near cyclin D1 in mouse lymphomas: A parallel for BCL1 translocations in human B-cell neoplasms. Oncogene 7:2381, 1992 |
|
Swerdlow SH, Yang WI, et al: The morphologic spectrum of non-Hodgkin's lymphomas with BCL1/cyclin D1 gene rearrangements. Am J Surg Pathol 20:627, 1996 |
|
Guerts JM, Roijer E, et al: Expression of reciprocal hybrid transcripts of HMGIC and FHIT in a pleomorphic adenoma of the parotid gland. Cancer Res 57:13, 1997 |
|
Kazmierczak B, et al: Cloning and molecular characterization of part of a new gene fused to HMGIC in mesenchymal tumors. Am J Pathol 152:431-435, 1998 |
|
Kazmierczak B, et al: Description of a novel fusion transcript between HMGI-C, a gene encoding for a member of the high mobility group proteins, and the mitochondrial aldehyde dehydrogenase gene. Cancer Res 55:6038-6039, 1995 |
|
Quade BJ, Weremowicz S, Neskey DM, et al: Fusion Transcripts Involving HMGA2 Are not a Common Molecular Mechanism in Uterine Leiomyomata with Rearrangements in 12q15. Cancer Res 63:(6):1351-1358, 2003 |
|
Winkler VDH, Hoffmann W: Regarding the question of inheritance of uterine myoma. Deutsche-Medizinische Wochenschrift 68:(8):235-257, 1938 |
|
Faerstein E, Szklo M, Rosenshein NB: Risk factors for uterine leiomyoma: A practice-based case-control study. II. Atherogenic risk factors and potential sources of uterine irritation Am J Epidemiol 153:(1):11-19, 2001 |
|
Marshall LM, et al: Variation in the incidence of uterine leiomyomata among premenopausal women by age and race. Obstet Gynecol 90:967-973, 1997 |
|
Treloar SA, et al: Pathways to hysterectomy: Insights from longitudinal twin research. Am J Obstet Gynecol 167:(1):82-88, 1992 |
|
Kurbanova M, Koroleva AG, Sergeev AS: Genetic analysis of the structure of the predisposition to uterine myoma: prevalence and incidence. Genetika 25:(6):1122-1124, 1989 |
|
Kurbanova M, Koroleva AG, Sergeev AS: Genetic-epidemiological analysis of uterine myoma: estimate of risk to relatives. Genetika 20:(10):1896-1898, 1989 |
|
Vikhlyaeva EM, Khodzhaeva ZS, Fantschenko ND: Familial predisposition to uterine leiomyomas. Int J Gynecol Obstet 51:127-131, 1995 |
|
Schwartz S, et al: Familial aggregation of uterine leiomyomata. Presented at the annual meeting of the Society for Epidemiological Research. Seattle, WA, June 2000 |
|
Luoto R, et al: Heritability and risk factors of uterine fibroids--the Finnish Twin Cohort study. Maturitas 37:(1):15-26, 2000 |
|
Snieder H, MacGregor AJ, Spector TD: Genes control the cessation of a woman's reproductive life: A twin study of hysterectomy and age at menopause. J Clin Endocrinol Metab 83:(6):1875-1880, 1998 |
|
Reed WB, Walker R, Horowitz R: Cutaneous leiomyomata with uterine leiomyomata. Acta Dermatovener (Stockholm) 53:409-416, 1973 |
|
Launonen V, et al: Inherited suseptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 98:(6):3387-3392, 2001 |
|
Thyresson HN, Su WPD: Familial cutaneous leiomyomatosis. J Am Acad Dermatol 4:430-434, 1981 |
|
Astuti D, et al: Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69:(1):49-54, 2001 |