Preimplantation Diagnosis for Mendelian Disorders
Authors
INTRODUCTION
PGD (preimplantation genetic diagnosis) has become an established procedure for avoiding the birth of affected children with single gene disorders and having a healthy unaffected offspring of their own. PGD is performed through polar body or embryo biopsy, which have no deleterious effect on the pre- and post-implantation development. The review describes recent developments in improving the accuracy of PGD for Mendelian disorders, and current changes in the spectrum of conditions for which PGD has been applied. Among the most recent applications of PGD were PGD for congenital malformations, blood group incompatibility, and increasing numbers of late onset disorders with genetic predisposition, which have never been indications for prenatal diagnosis. Despite ethical concerns, PGD has been further applied for preselection of unaffected and HLA matched embryos, and also for preimplantation HLA matching without testing for causative gene. This extends the practical value of PGD, with its utility being no longer limited to prevention of single gene disorders, but expanded to treatment of siblings requiring stem cell transplantation.
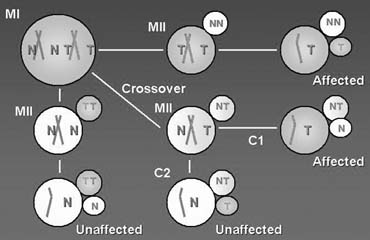
PGD is a novel approach to detect genetic disorders before pregnancy, avoiding the need for prenatal diagnosis and abortion. It is based on testing of oocytes or embryos to pre-select and transfer only normal embryos back to the patient, achieving an unaffected pregnancy and birth of a healthy child. PGD is performed either by testing single blastomeres or a few trophectoderm cells removed from preimplantation embryos, or by testing female gametes (Fig. 1).1, 2 The latter is based on the removal and genetic analysis of the first and second polar bodies (PB1 and PB2), which represent the byproducts of the first and second meiotic divisions (see Fig. 1A, 1B, 1C). Because neither PB1 nor PB2 have any biologic significance for embryo development, they may be removed and tested for the genetic contents to predict the resulting maternal contribution to the embryo. However, this approach cannot be applied for testing of paternally derived abnormalities, and gender determination, which may be detected by genetic analysis of single blastomeres or trophectoderm cells, removed from preimplantation embryos (see Fig. 1D). Both methods, therefore, are complementary and may be applied depending on the PGD objectives in each patient.
|
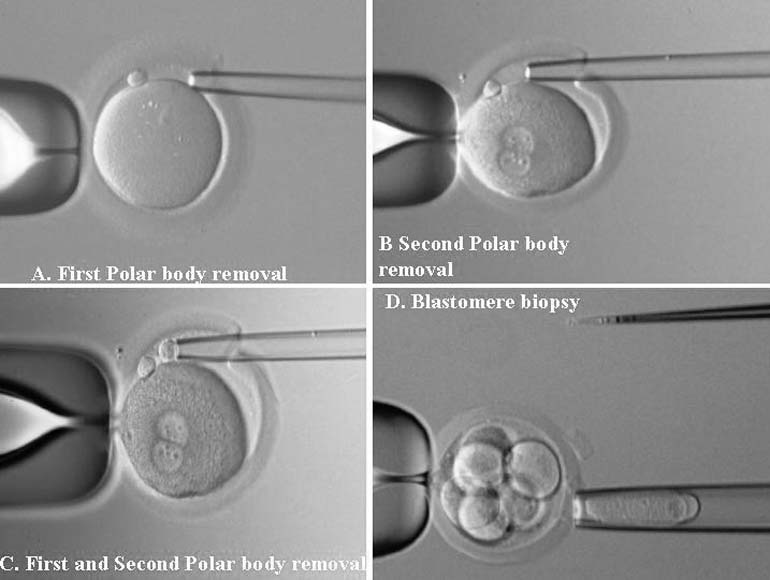
More than 30,000 PGD cycles have been performed and resulted in the birth of thousands of children following the procedure.3, 4 The overall experience of using PGD for single gene disorders is presently almost 1500 cases, showing that it is an established alternative to traditional prenatal diagnosis, which may be reliably applied as an integral part of genetic practices.4, 5, 6, 7, 8 As will be demonstrated, PGD has appeared to be applicable to a wider range of inherited conditions which have never been indications for prenatal diagnosis, and also made it possible to initiate nondisease testing to preselect HLA-compatible offspring in addition to PGD for causative gene, which makes it possible to facilitate treatment of siblings requiring stem cell transplantation.
GENETIC ANALYSIS IN SINGLE CELLS AND AVOIDING MISDIAGNOSIS
Sources for Misdiagnosis
Because PGD for single gene disorders is based on single cell genetic analysis, its accuracy largely depends on the limitations of single cell DNA analysis, which may potentially cause misdiagnosis (Table 1). One of the key contributors to misdiagnosis is the phenomenon of preferential amplification, also known as allele-specific amplification failure (allele drop out-ADO), requiring the application of special protocols to ensure the highest ADO detection rate (Fig. 2).1, 2, 9 A A few previously reported misdiagnoses, involving PGD for beta-thalassemia, myotonic dystrophy (DM), fragile-X syndrome (XMR1) and cystic fibrosis (CF), might have been due to this phenomenon, which has not initially been fully realized.5, 7
Table 1. Problems of PGD for single gene disorders
| Prevention and elimination of DNA contamination |
| Optimization of reaction conditions |
| Detection of allele drop-out (ADO) and preferential amplification |
| Detection of aneuploidy |
{kind=link}
It has been demonstrated that ADO rates in single cells might be different for different types of heterozygous cells.2, 10 The ADO rate may exceed 20% in blastomeres compared with ADO rate in single fibroblasts and PB1, which was shown to be less than 10% (Fig. 3). A high rate of ADO in blastomeres may lead to an obvious misdiagnosis, especially in compound heterozygous embryos. As mentioned, most misdiagnoses, especially those at the initial stage of application of PGD for single gene disorders, were in the cases of blastomere biopsy from apparently compound heterozygous embryos.
{kind=link}
Use of Polymorphic Markers
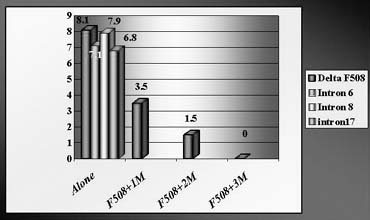
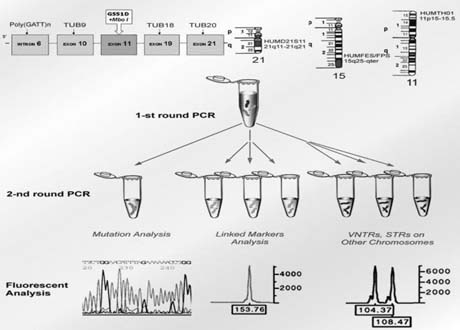
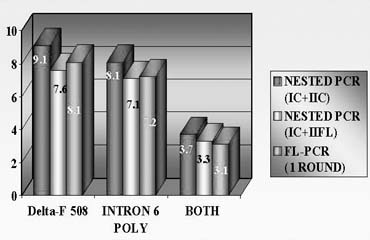
To avoid a misdiagnosis caused by preferential amplification, a simultaneous detection of mutant gene together with up to three highly polymorphic markers closely linked to the gene tested was introduced.1, 2, 9 Each additional linked marker reduced misdiagnosis rate approximately by half, so with one linked marker amplified together with mutation, a misdiagnosis risk in blastomere analysis may be reduced from 20% to 10%, with two from 10% to 5% and with three from 5% to practically zero (Fig. 4). So, a multiplex nested polymerase chain reaction (PCR) analysis is performed, with the initial PCR containing all the pairs of outside primers, so that after the first-round PCR, separate aliquots of the resulting PCR product may be amplified using the inside primers specific for each site (Fig. 5). Only when the polymorphic sites and the mutation agree are embryos transferred. So, multiplex amplification allows detecting of ADO and prevents the transfer of misdiagnosed affected embryos.
{kind=link}
{kind=link}
Sequential PB1 and PB2 DNA Analysis
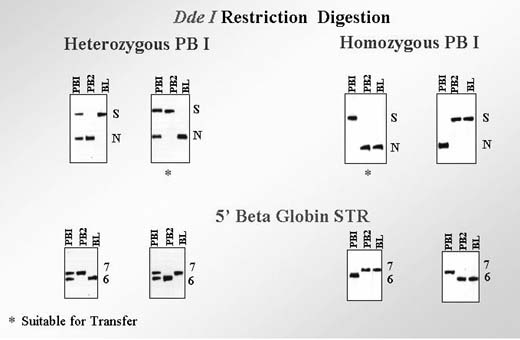
Another efficient approach for avoiding misdiagnosis may be a sequential genetic analysis of the PB1 and PB2 in PGD for maternally derived mutations.1, 2 Detection of both mutant and normal alleles in the heterozygous PB1, together with the mutant allele in the corresponding PB2, leaves no doubt that the resulting maternal contribution to the embryo is normal, even without testing for the linked markers as a control (Fig. 6). However, it will be ideal to test simultaneously at least for one linked marker to confirm the diagnosis.
{kind=link}
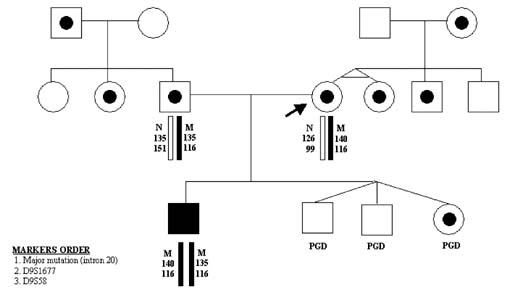
Alternatively, the mutation-free oocytes may be also predicted when corresponding PB1 is homozygous mutant (Fig. 7). In that scenario, the corresponding PB2 should be hemizygous-normal, similar to the resulting maternal pronucleus. However, the genotype of the resulting maternal contribution may be quite opposite, in other words, mutant, if the corresponding PB1 is in fact heterozygous but erroneously diagnosed as homozygous-normal because of ADO of the normal allele. In the aforementioned scenario, the extrusion of the normal allele with PB2 would lead to the mutant allele left in the resulting oocyte. Therefore, the embryos resulting from the oocytes with homozygous mutant PB1 cannot be acceptable for transfer, unless the heterozygous status of PB1 is excluded by the use of linked markers as described. The example of misdiagnosis, caused by ADO of the normal allele in PB1 has been described earlier in a PGD cycle performed for FMR1 (Table 2).11 To completely avoid misdiagnosis, a sequential PB1 and PB2 may be required to combine with multiplex PCR to exclude the possibility of an undetected ADO in heterozygous PB1. The analysis of more than 1000 oocytes tested by sequential PB1 and PB2 analyses showed that more than half of ADOs were detected by sequential analysis of PB1 and PB2, with the remaining cases detected by multiplex PCR.9 The accuracy of this approach may be demonstrated by the reports of PGD for thalassemia and familial dysautonomia (FD), resulting in the transfer of three unaffected embryos in each case, which were confirmed by the birth of the two sets of triplets free from thalassemia and FD (Fig. 8).12, 13
{kind=link}
Table 2. PGD for FMRI gene by linkage analysis in case of misdiagnosis
| Oocyte # | Cell Type | DXs1193 (in bp) | DXs548 (in bp) | Predicted Oocyte Genotype |
| 1 | PB1 | 154*/156 | 245/247 | Affected |
| PB2 | 156 | 247 | ||
| 3 | PB1 | 154/156 | 245/247 | Affected |
| PB2 | 156 | 247 | ||
| 5 | PB1 | 154/156 | 245/247 | Normal |
| PB2 | 154 | 245 | ||
| 6 | PB1 | 154/156 | 245/247 | Normal |
| PB2 | 154 | 245 | ||
| 9 | PB1 | 154 | 245 | Normal† |
| PB2 | 156 | 247 | ||
| 10 | PB1 | 154/156 | 245/247 | Affected |
| PB2 | 156 | 247 | ||
| Cord blood | 154 | 245 | Affected |
PB 1 first polar body; PB 2 second polar body; Bp, size of PCR product in base pairs.
*Bold figures show markers linked to the expanded allele.
†Potential 5% risk for ADO (allele drop-out)
{kind=link}
Fluorescent and Real Time PCR
The other method with the proved potential for detecting and avoiding misdiagnosis caused by preferential amplification is fluorescence PCR (F-PCR), which may allow detection of some of the heterozygous PB1 or blastomeres misdiagnosed as homozygous in conventional PCR, therefore having potential of reducing the ADO rates at least to some extent (Fig. 9).9 In addition, the method also allows a simultaneous gender determination, DNA fingerprinting, and detection of common aneuploidies. With further improvement and simplification, F-PCR combined with a multiplex system and sequential PB1 and PB2 analysis in cases of maternally derived mutations may allow excluding almost completely the risk for misdiagnosis caused by preferential amplification.
{kind=link}
The accuracy of PGD has been further improved with the application of fluorescent PCR with the expand long template (ELT) kit, which enabled reduction of the ADO rate from as high as 30–35% in both conventional and fluorescent PCR to as low as 5% in testing for DM.14 Another development in improving the accuracy of single cell PCR analysis involves the application of real-time PCR, which was found to reduce the ADO rate almost by half, compared with conventional or fluorescent PCR2. The application of these approaches together with simultaneous testing for the causative mutation along with at least one or two linked markers may allow avoiding reliably the risk for misdiagnosis.
Simultaneous Testing for Aneuploidy
Finally, because of high rate of mosaicism at the cleavage stage, testing for the chromosome, in which the gene in question is mapped, is of an obvious value to exclude the lack of mutant allele caused by monosomy of this chromosome in the biopsied blastomere. As mentioned, aneuploidy testing is technically feasible and may be performed by adding primers for chromosome-specific microsatellite markers to the multiplex PCR protocols worked out for specific genetic disorders.15 The development of multiplex nested PCR systems will also allow performing PGD for different conditions simultaneously, as attempted for CFTR mutation together with XMR1 or gender determination, for two different conditions when the couples are at risk for producing the offspring with more than one genetic disease.16, 17, 18, 19 Simultaneous detection of different genetic conditions and aneuploidy may in future become routine with the progress of single cell microarray technology.20
Custom-Made PGD Design
Because of the need for the development of a custom-made PGD design for each mutation and each couple, a preparatory work has become an integral part of PGD for single gene disorders to ensure avoiding the potential misdiagnosis. For example, in some cases a particular set of outside primers has to be designed to eliminate false priming to the pseudogene, as described in PGD for long-chain 3-hydroxyacyl-Coa dehydrogenase deficiency (Fig. 10).21 Also, the preparatory work may frequently involve a single sperm-typing needed for establishing paternal haplotypes so that linked marker analysis could be performed in addition to mutation analysis, especially in cases of paternally derived dominant conditions or PGD combined with preimplantation HLA-matching (see later). The availability of the parental haplotypes allows not only the confirmation of the absence of the mutant gene but also the presence of both maternal and paternal wild alleles in PGD by blastomere analysis, especially when only one informative marker is available, as demonstrated in PGD for sonic hedgehog mutation (Table 3).22
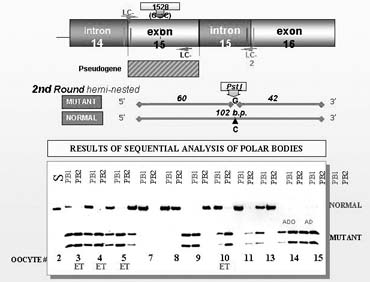{kind=link}
Table 3. Results of multiplex PCR analysis with simultaneous testing for SHH mutation and linked marker (D7S550) and the follow up embryo analysis for confirmation of single blastomere diagnosis
| Embryo # | Mutation Analysis (SHH gene) | Linked Marker Analysis (D7S550)* | Predicted Genotype | Transfer/Follow-Up Analysis |
| 2 | N/ADO | 2/4 | Carrier (affected) | Confirmed |
| 4 | N/N | 1/3 | Normal | Transferred |
| 5 | N/N | 1/3 | Normal | Transferred |
| 8 | N/N | 2/3 | Normal | Frozen |
| 9 | N/ADO | 2/4 | Carrier (affected) | Confirmed |
| 10 | ADO/M | 1/4 | Carrier (affected) | Confirmed |
| 16 | N/N | 2/3 | Normal | Frozen |
| 17 | N/ADO | 1/4 | Carrier (affected) | Confirmed |
| 19 | N/ADO | 1/4 | Carrier (affected) | Confirmed |
| Mother | N/N | 1/2 | Normal | |
| Father | N/M | 3/4 | Carrier (mosaic) |
N, normal allele; M, mutant allele; ADO, allele dropout.
*Linked markers:
Maternal PCR product 138 base pairs = 1
Maternal PCR product 158 base pairs = 2
Paternal PCR product 152 base pairs = 3 (linked to normal gene)
Paternal PCR product 156 base pairs = 4 (linked to mutant gene)
EXPANDING PGD APPLICATION
The list of disorders, presently comprising more than 170 different conditions, for which PGD was applied is being extended beyond the indications for prenatal diagnosis, although the most frequent ones are still CF and hemoglobin disorders (Table 4).5, 7, 23, 24 According to our experience of over thousand PGD cycles for single gene disorders, almost half of these cycles were performed for CF and hemoglobin disorders, followed by DM and XMR1,25 similar to the experiences of the other active centers.7 There have been many previous reports on PGD for different single gene disorders as well extensive reviews on the subject.3, 9, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31 So we will present only the most recent developments and the expanding application of PGD, which has been shown to be useful for much wider indications than those used in traditional prenatal diagnosis.
Table 4 List of diseases and genes for which PGD has been performed
| Disease | MIM number | Inheritance | Gene name/symbol | Protein name | Location |
| Achondroplasia; ACH | 100800 | 100800 | FGFR3 | Fibroblast growth factor receptor 3 [precursor] | 4 p16.3 |
| Acyl-CoA dehydrogenase, medium-chain, deficiency | 201450 | AR | ACADM | Acyl-CoA dehydrogenase, medium-chain specific, mitochondrial [precursor] | 1p31 |
| Acyl-CoA dehydrogenase, very long-chain; ACADVL | 609575 | AR | ACADVL | Acyl-coenzyme A dehydrogenase, very long chain | 17p13-p11 |
| Adadenosine deaminase deficiency; ADA | 102700 | AR | ADA | Adenosine deaminase | 20q13.11 |
| Adenomatous polyposis of the colon; APC | 175100 | AD | APC | Adenomatous polyposis coli protein | 5 q21-q22 |
| Adrenoleukodystrophy; ALD | 300100 | XL | ABCD1 | Adrenoleukodystrophy protein | Xq28 |
| Albinism, ocular, type I; OA1 | 300500 | XL | OA1 | G-protein coupled receptor 143 | Xp22.3 |
| Alopecia universalis congenita; ALUNC | 203655 | AR | HR | Hairless protein | 8 p21.2 |
| Alpers diffuse degeneration of cerebral gray matter with hepatic cirrhosis | 203700 | AR | POLG | Mitochondrial DNA polymerase gamma | 15q25 |
| Alpha 1 antitrypsin deficiency (AAT) | 107400 | AR | SERPINA1 | Alpha-1-antitrypsin [precursor] | 14q32.1 |
| Alport syndrome, X-linked; ATS | 301050 | XL | AMMECR1 | AMME syndrome candidate gene 1 protein | Xq22.3 |
| Amyloidosis I, hereditary neuropathic | 176300 | AD | TTR | Transthyretin [precursor] | 18q11.2-q12.1 |
| Androgen receptor; AR (testicular feminization; spinal and bulbar muscular atrophy; Kennedy disease) | 313700 | XL | AR | AR protein | Xq11-q12 |
| Aneuploidies by STR genotyping | | ||||
| Angioedema, hereditary; HAE | 106100 | AD | SERPING1 | Plasma protease C1 inhibitor precursor | 11q11-q13.1 |
| Ataxia-telangiectasia; AT | 208900 | AR | ATM | Serine-protein kinase ATM | 11q22-q23 |
| Basal cell nevus syndrome; BCNS (Gorlin) | 109400 | AD | PTCH | Patched protein homolog 1 | 9q22.1–31 |
| Blepharophimosis, ptosis, and epicanthus inversus; BPES | 110100 | AD | FOXL2 | Forkhead box protein L2 | 3 q23 |
| Blood group − Kell−Cellano system | 110900 | AD | KEL | Kell blood group glycoprotein | 7 q33 |
| Brachydactyly, Type B1; BDB1 | 113000 | AR | ROR2 | Receptor tyrosine kinase-like orphan receptor 2 | 9q22 |
| Brain tumor, posterior fossa of infancy, familial | 601607 | AD | SMARCB1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily B member | 22q11.2 |
| Breast cancer, familial | 113705 | AD | BRCA1 | Breast cancer type 1 susceptibility protein | 17q21 |
| Breast cancer, familial | 600185 | AD | BRCA2 | Breast cancer type 2 susceptibility protein | 13q12.3 |
| Bruton agammaglobulinaemia tyrosine kinase; BTK | 300300 | XL | BTK | Bruton agammaglobulinaemia tyrosine kinase | Xq21.33-q22 |
| Canavan disease | 271900 | AR | ASPA | Aspartoacylase | 17pter-p13 |
| Ceroid lipofuscinosis, neuronal 2, late infantile; CLN2 | 204500 | AR | CLN2 | Tripeptidyl-peptidase I [precursor] | 11p15 |
| Charcot−Marie−Tooth disease, axonal, type 2E | 607684 | AD | NEFL | Neurofilament triplet L protein | 8 p21 |
| Charcot−Marie−Tooth disease, demyelinating, type 1A; CMT1A | 118220 | AD | PMP22 | Peripheral myelin protein 22 | 17p12 |
| Charcot-Marie-Tooth disease, demyelinating, type 1B; CMT1B | 118200 | AD | MPZ | Myelin P0 protein [precursor] | 1q23.3 |
| Charcot−Marie−Tooth disease, X-linked, 1; CMTX1 | 302800 | XL | GJB1 | Gap junction beta-1 protein | Xq13.1 |
| Cholestasis, progressive familial intrahepatic 2 | 603201 | AR | ABCB11 | ATP-binding cassette, sub-family B (MDR/TAP), member 11 | 2q24 |
| Chondrodysplasia punctata 1, X-linked recessive; CDPX1 | 302950 | XL | ARSE | Arylsulfatase E | Xp22.3 |
| Choroideraemia; CHM | 303100 | XL | CHM | Rab proteins geranylgeranyltransferase component A 1 | Xq21.2 |
| Citrullinaemia, classic | 215700 | AR | ASS | Argininosuccinate synthase | 9q34.1 |
| Collagen, type IV, alpha-5; COL4A5 | 303630 | XL | COL4A5 | Collagen, type IV, alpha 5 | Xq22.3 |
| Colorectal cancer, hereditary non-polyposis, type 1; HNPCC1 | 120435 | AD | MSH2 | DNA mismatch repair protein Msh2 | 2p 2-p21 |
| Colorectal cancer, hereditary non-polyposis, type 2; HNPCC2 | 609310 | AD | MLH1 | DNA mismatch repair protein Mlh1 | 3 p21.3 |
| Congenital adrenal hyperplasia (CAH) | 201910 | AR | CYP21A2 | Cytochrome P450 XXIB | 6 p21.3 |
| Craniofacial dysostosis, type I; (CFD1) | 123500 | AD | FGFR2 | Fibroblast growth factor receptor 2 [precursor] | 10q26.13 |
| Currarino syndrome | 176450 | AD | HLXB9 | Homeobox protein HB9 | 7q36 |
| Cutis laxa, autosomal recessive, type I | 219100 | AR | FBLN4 | EGF-containing fibulin-like extracellular matrix protein 2 | 11q13 |
| Cystic fibrosis; CF | 219700 | AR | CFTR | Cystic fibrosis transmembrane conductance regulator | 7q31.2 |
| Cystinosis, nephropathic; CTNS | 219800 | AR | CTNS | Cystinosin | 17p13 |
| Darier−White disease; DAR | 124200 | AD | ATP2A2 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | 12q23-q24.1 |
| Deafness, neurosensory, autosomal recessive 1; DFNB1 | 220290 | AR | GJB2 | Gap junction protein connexin-26 | 13q11-q12 |
| Diamond−Blackfan anaemia; DBA | 105650 | AD | RPS19 | 40S ribosomal protein S19 | 19q13.2 |
| Dysautonomia, familial | 223900 | AR | IKBKAP | 9q31 | |
| Dystrophia myotonica 1 | 160900 | AD | DMPK | Myotonin-protein kinase | 19q13.2-q13.3 |
| Early-onset familial Alzheimer disease | 104760 | AD | APP | Amyloid beta A4 protein [precursor] | 21q21.3 |
| Ectodermal dysplasia 1, anhidrotic; ED1 | 305100 | XL | ED1 | Ectodysplasin A | Xq13.1 |
| Ectodermal dysplasia, anhidrotic | 224900 | AR | EDAR | Tumor necrosis factor receptor superfamily member EDAR [precursor] | 2q 11-q13 |
| Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 1; EEC1 | 129900 | AD | p63 | Tumor protein 63 | 7 q11.2-q21.3 |
| Emery−Dreifuss muscular dystrophy, autosomal recessive; EDMD3 | 604929 | AR | LMNA | Lamin A/C | 1q21.2 |
| Emery−Dreifuss muscular dystrophy, X-linked; EDMD | 310300 | XL | EMD | Emerin | Xq28 |
| Epidermolysis bullosa dystrophica, Pasini type | 131750 | AR | COL7A1 | Collagen alpha 1(VII) chain [precursor] | 3 p21.3 |
| Epidermolysis bullosa lethalis | 226650 | AR | LAMB3 | Laminin, beta 3 | 1q32 |
| Epidermolysis bullosa simplex and limb-girdle muscular dystrophy | 226670 | AR | PLEC1 | Plectin 1, intermediate filament binding protein 500kDa | 8q24 |
| Epiphyseal dysplasia, multiple, 1; EDM1 | 132400 | AD | COMP | Cartilage oligomeric matrix protein [precursor] | 19p13.1 |
| Exostoses, multiple, type I | 133700 | AD | EXT1 | Exostosin-1 | 8 q24.11q24.13 |
| Fabry disease | 301500 | XL | GLA | Alpha-galactosidase A [precursor] | Xq22 |
| Facioscapulohumeral muscular dystrophy 1A; FSHMD1A | 158900 | AD | FRG1 | FRG1 protein | 4 q35 |
| Fanconi anaemia, complementation group C; FANCC | 227645 | AR | FANCC | Fanconi anaemia group C protein | 9q22.3 |
| Fanconi anaemia, complementation group E; FANCE | 600901 | AR | FANCE | Fanconi anaemia, complementation group E | 6p22-p21 |
| Fanconi anaemia, complementation group F; FANCF | 603467 | AR | FANCF | Fanconi anaemia group F protein | 11p15 |
| Fanconi anaemia, complementation group G | 602956 | AR | XRCC9 | DNA-repair protein XRCC9 | 9p13 |
| Fanconi anaemia, complementation group J | 609054 | AR | BRIP1 | Fanconi anaemia group J protein | 17q22 |
| Fanconi anaemia, complementation group A; FANCA | 227650 | AR | FANCA | Fanconi anaemia group A protein | 16q24.3 |
| Fragile site mental retardation 1 | 309550 | XL | FMR1 | Fragile X mental retardation 1 protein | Xq27.3 |
| Fragile site, folic acid type, rare, FRA(X)(q28); FRAXE | 309548 | XL | FMR2 | Fragile X mental retardation 2 protein | Xq28 |
| Friedreich ataxia 1; FRDA | 229300 | AR | FRDA | Frataxin, mitochondrial precursor | 9q13 |
| Galactosaemia | 230400 | AR | GALT | Galactose-1-phosphate uridylyltransferase | 9p13 |
| Gangliosidosis, generalized GM1, type I | 230500 | AR | GLB1 | Galactosidase, beta 1 | 3p21.33 |
| Gaucher disease, type I | 230800 | AR | GBA | Glucosylceramidase [precursor] | 1q21 |
| Glomuvenous malformation (GVM) | 138000 | AD | TIE2/TEK | 1p22-p21 | |
| Glutaric acidaemia I | 231670 | AR | GCDH | Glutaryl-coenzyme A dehydrogenase | 19p13.2 |
| Glycogen storage disease type VI | 232700 | AR | PYGL | Glycogen phosphorylase, liver form | 14q21-q22 |
| Haemoglobin-alpha locus 1; HBA1 | 141800 | AR | HBA1 | Haemoglobin alpha chain | 16pter-p13.3 |
| Haemoglobin-alpha locus 2; HBA2 | 141850 | AR | HBA2 | Haemoglobin alpha subunit | 16pter-p13.3 |
| Haemoglobin-beta locus; HBB | 141900 | AR | HBB | Haemoglobin beta chain | 11p15.5 |
| Hemophagocytic lymphohistiocytosis, familial, 2 | 603553 | AR | PRF1 | Perforin 1 [precursor] | 10q22 |
| Hemophilia A | 306700 | XL | F8 | Coagulation factor VIII [precursor] | Xq28 |
| Hemophilia B | 306900 | XL | F9 | Coagulation factor IX [precursor] | Xq27.1-q27.2 |
| HLA matching genotyping | 6 | ||||
Homocystinuria due to deficiency of N(5,10)-methylene tetrahydrofolate reductase activity | 236250 | AR | MTHFR | Methylenetetrahydrofolate reductase | 1p36.3 |
| Hoyeraal−Hreidarsson syndrome; HHS | 300240 | XL | DKC1 | H/ACA ribonucleoprotein complex subunit 4 | Xq28 |
| Huntington disease; HD | 143100 | AD | HD | Huntingtin | 4 p16.3 |
| Hurler syndrome | 607014 | AR | IDUA | Alpha-L-iduronidase [precursor] | 4 p16.3 |
| Hydrocephalus, X-linked; L1CAM | 308840 | XL | L1CAM | Neural cell adhesion molecule L1 [precursor] | Xq28 |
| Hyperinsulinemic hypoglycaemia, familial, 1; HHF1 | 256450 | AR | ABCC8 | ATP-binding cassette, sub-family C (CFTR/MRP), member 8 | 11p15.1 |
| Hypophosphatasia, infantile | 241500 | AR | ALPL | Alkaline phosphatase, tissue-non-specific isozyme [precursor] | 1p36.1–34 |
| Hypophosphatemic rickets, X-linked dominant | 307800 | XL | PHEX | Phosphate regulating endopeptidase homolog | Xp22.2-p22.1 |
| Hypophosphatemic rickets, X-linked dominant | 307800 | XL | PHEX | Phosphate regulating endopeptidase homolog | Xp22.2-p22.1 |
| Immunodeficiency with hyper-IgM, type 1; HIGM1 | 308230 | XL | TNFSF5 | Tumor necrosis factor ligand superfamily member 5 | Xq26 |
| Incontinentia pigmenti; IP | 308300 | XL | IKBKG | NF-kappa-B essential modulator | Xq28 |
| Isovaleric acidaemia; IVA | 243500 | AR | IVD | Isovaleryl coenzyme A dehydrogenase | 15q14-q15 |
| Krabbe disease | 245200 | AR | GALC | Galactocerebrosidase [precursor] | 14q31 |
| Leigh syndrome; LS | 185620 | AR | SURF1 | Surfeit locus protein 1 | 9q34.2 |
| Leukoencephalopathy with vanishing white matter; VWM | 603896 | AR | EIF2B2 | Translation initiation factor eIF-2B beta subunit | 14q24 |
| Li−Fraumeni syndrome 1; LFS1 | 151623 | AD | TP53 | Cellular tumor antigen p53 | 17p13.1 |
| Loeys−Dietz syndrome; LDS | 609192 | AD | TGFBR2 | Transforming growth factor, beta receptor II (70/80kDa) | 3p22 |
| Long-chain 3-hydroxyacyl-coa dehydrogenase deficiency;HADHA | 600890 | AR | HADHA | Trifunctional enzyme alpha subunit, mitochondrial [precursor] | 2p 3 |
| Machado−Joseph disease; MJD | 109150 | AD | ATX3 | Machado−Joseph disease protein 1 | 14q24.3-q31 |
| Marfan syndrome; MFS | 154700 | AD | FBN1 | Fibrillin 1 [precursor] | 15q21.1 |
| Meckle-Gruber Syndrome | 249000 | AR | MKS1 | 17q23 | |
| Metachromatic leukodystropy | 250100 | AR | ARSA | Arylsulfatase A [precursor] | 22q13.31-qter |
| Metaphyseal chondrodysplasia, schmid type; MCDS | 156500 | AD | COL10A1 | Collagen, type X, alpha 1 | 6q21-q22 |
| Methylmalonic aciduria and homocystinuria (MMACHC) | 277400 | AR | MMACHC | 1p34.1 | |
| Microcoria-congenital nephrosis syndrome | 609049 | AR | LAMB2 | Laminin beta-2 | 3p21 |
| Migraine, familial hemiplegic, 1; FHM1 | 141500 | AD | CACNA1A | Calcium channel, voltage-dependent, P/Q type, alpha 1A subunit | 19p13.2-p13.1 |
| Morquio syndrome, non-keratosulfate-excreting type | 252300 | AR | GALNS | Galactosamine (N-acetyl)-6-sulfate sulfatase | 16q24.3 |
| Mucopolysaccharidosis type II (Hunter) Hunter−McAlpine craniosynostosis syndrome | 309900 | AD | IDS | Iduronate 2-sulfatase [precursor] | Xq28 |
| Multiple acyl-CoA dehydrogenase deficiency; MADD | 231680 | AR | ETFA | Electron-transfer-flavoprotein, alpha polypeptide | 15q23-q25 |
| Multiple endocrine neoplasia, type I; MEN1 | 131100 | AD | MEN1 | Multiple endocrine neoplasia I | 11q13.1 |
| Multiple endocrine neoplasia, type IIA; MEN2A | 171400 | AD | RET | Ret proto-oncogene | 10q11.2 |
| Muscular dystrophy, Becker type; BMD | 300376 | XL | DMD | Dystrophin | Xq21.2 |
| Muscular dystrophy, Duchenne type; DMD | 310200 | XL | DMD | Dystrophin | Xq21.2 |
| Myotubular myopathy 1; MTM1 | 310400 | XL | MTM1 | Myotubularin | Xq28 |
| N-acetylglutamate synthase deficiency | 237310 | AR | NAGS | N-acetylglutamate synthase | 17q21.31 |
| Neurofibromatosis, type I; NF1 | 162200 | AD | NF1 | Neurofibromin | 17q11.2 |
| Neurofibromatosis, type II; NF2 | 101000 | AD | NF2 | Merlin | 22q12.2 |
| Neuropathy, hereditary sensory and autonomic, type III; HSAN3 | 223900 | AR | IKBKAP | Kinase complex-associated protein | 9q31 |
| Niemann-Pick Disease | 257220 | AR | NPC1 | 18q11-q12 | |
| Norrie disease; NDP | 310600 | XL | NDP | Norrin | Xp11.4-p11.3 |
| Oculocutaneous albinism, type I; OCA1 | 203100 | AR | TYR | Tyrosinase [precursor] | 11q14-q21 |
| Oculocutaneous albinism, type II; OCA2 | 203200 | AD | OCA2 | P protein | 15q11.2-q12 |
| Omenn syndrome | 603554 | AD | RAG1 | V(D)J recombination-activating protein 1 | 11p13 |
| Optic atrophy 1; OPA1 | 165500 | AD | OPA1 | Dynamin-like 120 kDa protein, mitochondrial [precursor] | 3 q28-q29 |
| Ornithine transcarbamylase deficiency | 311250 | XL | OTC | Ornithine carbamoyltransferase, mitochondrial [precursor] | Xp21.1 |
| Osteogenesis imperfecta congenita; OIC | 166200 | AD | COL1A2 | Collagen alpha 2(I) chain [precursor] | 7 q22.1 |
| Osteogenesis imperfecta congenita; OIC | 166200 | AD | COL1A1 | Collagen alpha 1(I) chain [precursor] | 17q21.31-q22 |
| Osteopetrosis, autosomal recessive | 259700 | AR | TCIRG1 | Vacuolar proton translocating ATPase 116 kDa subunit a isoform 3 | 11q13.4-q13.5. |
| Osteopetrosis, autosomal recessive | 259700 | AR | TCIRG1 | T-cell, immune regulator 1 | 11q13.2 |
| Pancreatitis, hereditary; PCTT | 167800 | AD | PRSS1 | Protease, serine, 1 (trypsin 1) | 7q32-qter|7q34 |
| Pelizaeus−Merzbacher-like disease; PMLD | 311601 | XL | PLP1 | Myelin proteolipid protein | Xq22 |
| Peutz−Jeghers syndrome; PJS | 175200 | AD | STK11 | Serine/threonine kinase 11 | 19p13.3 |
| Phenylketonuria | 261600 | AR | PAH | Phenylalanine-4-hydroxylase | 12q22-q24.2 |
| Polycystic kidney disease 1; PKD1 | 601313 | AD | PKD1 | Polycystin 1 precursor | 16P13.3 |
| Polycystic kidney disease 2; PKD2 | 173910 | AD | PKD2 | Polycystin 2 | 4 q22.1 |
| Polycystic kidney disease, autosomal recessive; ARPKD | 263200 | AR | PKHD1 | Polycystic kidney and hepatic disease 1 [precursor] | 6 p12.3 |
| Popliteal pterygium syndrome; PPS | 119500 | AD | IRF6 | Interferon regulatory factor 6 | 1q32-q41 |
| Propionic acidaemia | 232000 | AR | PCCA | Propionyl coenzyme A carboxylase, alpha polypeptide | 13q32 |
| Retinitis pigmentosa | 180380 | AD | RHO | Rhodopsin | 3q21-q24 |
| Retinitis pigmentosa 3; RP3 | 300389 | XL | RPGR | Retinitis pigmentosa GTPase regulator | Xp21.1 |
| Retinoblastoma; RB1 | 180200 | AD | RB1 | Retinoblastoma-associated protein | 13q14.1-q14.2 |
| Rett syndrome; RTT | 312750 | XL | MECP2 | Methyl-CpG-binding protein 2 | Xq28 |
| Rhesus blood group, CcEe antigens; RHCE | 111700 | AD | RHCE | CcEe antigens | 1p36.2-p34 |
| Rhesus blood group, D antigen; RHD | 111680 | AD | RHD | D antigen | 1p36.11 |
| Sandhoff disease | 268800 | AR | HEXB | Beta-hexosaminidase beta chain [precursor] | 5 q13 |
| Sickle cell anaemia | 603903 | AR | HBB | Haemoglobin beta chain | 11p15.5 |
| Smith−Lemli−Opitz syndrome; SLOS | 270400 | AR | DHCR7 | 7-Dehydrocholesterol reductase | 11q12-q13 |
| Sonic hedgehog; SHH | 600725 | AD | SHH | Sonic hedgehog protein [precursor] | 7 q36 |
| Spinal muscular atrophy, type I; SMA1 | 253300 | AD | SMN1 | Survival motor neuron protein | 5 q12.2-q13.3 |
| Spinocerebellar ataxia 1; SCA1 | 164400 | AD | ATXN1 | Ataxin 1 | 6p23 |
| Spinocerebellar ataxia 2; SCA2 | 183090 | AD | ATX2 | SCA2 protein | 12q24 |
| Spinocerebellar ataxia 6; SCA6 | 183086 | AD | CACNA1A | Voltage-dependent P/Q-type calcium channel alpha-1A subunit | 19p13 |
| Spinocerebellar ataxia 7; SCA7 | 164500 | AD | SCA7 | Ataxin-7 | 3 p21.1-p12 |
| Stickler syndrome, type I; STL1 | 108300 | AD | COL2A1 | Collagen, type II, alpha 1 | 12q13.11-q13.2 |
| Succinic semialdehyde dehydrogenase deficiency | 271980 | AR | ALDH5A1 | Succinate semialdehyde dehydrogenase, mitochondrial [precursor] | 6 p22 |
| Symphalangism, proximal; SYM1 | 185800 | AD | NOG | Noggin [precursor] | 17q22 |
| Tay-Sachs disease; TSD | 272800 | AR | HEXA | Beta-hexosaminidase alpha chain [precursor] | 15q23-q24 |
| Torsion dystonia 1, autosomal dominant; DYT1 | 128100 | AD | DYT1 | Torsin A [precursor] | 9q34 |
| Treacher Collins−Franceschetti syndrome; TCOF | 154500 | AD | TCOF | Treacle protein | 5 q32-q33.1 |
| Tuberous sclerosis type 1 | 191100 | AD | TSC1 | Hamartin | 9q34 |
| Tuberous sclerosis type 2 | 191100 | AD | TSC2 | Tuberin | 16p13.3 |
| Tyrosinaemia, type I | 276700 | AR | FAH | Fumarylacetoacetate hydrolase (fumarylacetoacetase) | 15q23-q25 |
| Ulnar−mammary syndrome; UMS | 181450 | AD | TBX3 | T-box 3 | 12q24.1 |
| Von Hippel−Lindau syndrome; VHL | 193300 | AD | VHL | Von Hippel−Lindau disease tumor suppressor | 3 p26-p25 |
| Wiskott−Aldrich Syndrome; WAS | 301000 | XL | WAS | Wiskott−Aldrich syndrome protein | Xp11.23-p11.22 |
| Zellweger syndrome; ZS | 214100 | AR | PXMP3 | Peroxisomal membrane protein 3, 35kDa | 8q21.1 |
| Zellweger syndrome; ZS | 214100 | AR | PEX1 | Peroxisome biogenesis factor 1 | 7 q21-q22 |
Specific Diagnosis for X-linked Diseases
More than one half of PGD cases for single gene disorders were performed by gender determination for X-linked conditions, which were the most straightforward application from the very beginning, either using PCR or FISH technique.5, 7 This was not only because the sequence information was not always available but also because it was technically easier to identify female embryos by DNA analysis or FISH technique, despite the obvious cost of discarding 50% healthy male embryos. However, testing for X-linked genetic disorders may be entirely limited to oocytes, because the mutations involved are fully maternally derived. Therefore, testing of oocytes for maternally derived specific mutations makes it possible to avoid further testing of the resulting embryos, which may be transferred irrespective of gender or any contribution from the father. Initially, the approach was applied for ornithine transcarbamylase deficiency (OTC)26 and then was extended to specific diagnosis of other X-linked disorders,11 and presently comprises the experience of specific diagnosis in a few dozens of clinical cycles performed for OTC, XMR1, myotubular myotonic dystrophy and X-linked hydrocephalus. This resulted in transfer of mutation free embryos in almost all cycles and yielding dozens unaffected clinical pregnancies. Specific diagnosis for X-linked mutations has also been performed at the cleavage stage.27, 28 The data demonstrate the clinical usefulness of the specific polar body or blastomere testing for X-linked disorders as an alternative to PGD by gender determination.
Couples with Homozygous Affected Partners
PGD was also provided for couples with one homozygous or double heterozygous-affected partner who were affected patients with thalassemia or phenylketonuria (PKU), resulting in an unaffected pregnancy and birth of healthy children.1, 2, 29 Although the risk for producing an affected child in such couples is as high as 50%, irrespective of maternal or paternal affected status, the strategy of PGD in such cases will depend on whether father or mother is affected. In couples with the affected fathers, PGD may concentrated on the preselection of mutation-free oocytes, while with the affected mothers, a cleavage stage PGD is required to identify those few embryos containing the normal gene.
The testing is particularly complicated if the parents are carrying different mutations. In one such case performed for thalassemia, the affected mother was double heterozygous (IVS I-110; IVS I-1-6) while the male partner was heterozygous carrier of the third mutation (IVS II-745).1, 25 This required a complex PGD design to exclude preferential amplification of each of the three alleles tested in blastomeres that underwent biopsy (Fig. 11), as also described in PGD for PKU.29 In this case, the affected father was compound heterozygous for R408 and Y414C mutations in exon 12 of phenylalanine hydroxylase (PAH) gene, and the carrier mother was heterozygous for R408W mutation in the same exon. PGD strategy was based on the preselection of the mutation-free oocytes using a sequential PB1 and PB2 DNA analysis. Based on the multiplex heminested PCR analysis, four embryos resulting from the zygotes predicted to contain no mutant allele of PHA gene were transferred, yielding an unaffected twin pregnancy and birth of the healthy twins (Fig. 12).
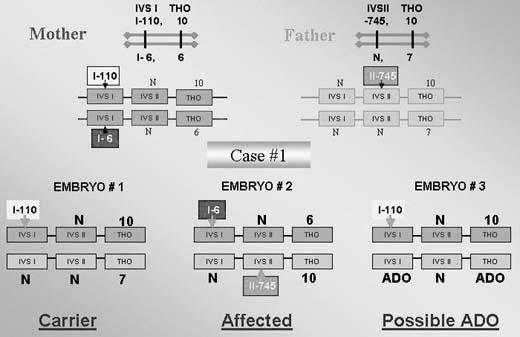{kind=link}
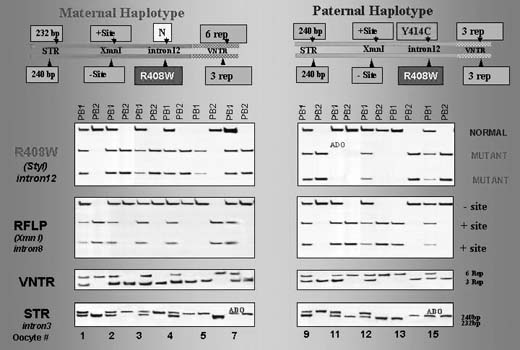{kind=link}
With the improvement of treatment and improved life expectancy for increasing number of genetic disorders, an increasing number of couples with affected maternal or paternal partners may require PGD as the only means for having unaffected children of their own.
Cancer predisposition has not traditionally been considered as an indication for prenatal diagnosis, because this would lead to pregnancy termination, which is not justified on the basis of genetic predisposition alone. However, the possibility of choosing embryos free of genetic predisposition for transfer would obviate the need for considering pregnancy termination, because only potentially normal pregnancies are established. PGD for such conditions appears acceptable on ethical grounds because only a limited number of the embryos available from hyperstimulation are selected for transfer.
The first PGD for inherited cancer predisposition have been performed for couples carrying p53 tumor-suppressor gene mutations known to determine a strong predisposition to the majority of cancers.30 The couple was with the paternally derived missense mutation caused by a transversion of a G-to-A in exon 5 of the p53 tumor-suppressor gene. The carrier was a 38-year-old proband with Li-Fraumeni syndrome (LFS), diagnosed with rhabdomyosarcoma of the right shoulder at the age of 2 followed by right upper extremity amputation. At the age of 31, he was also diagnosed with a high-grade leiomyosarcoma of the bladder and underwent a radical cystoprostatectomy. His mother was diagnosed with leiomyosarcoma at age 37.
PGD was performed by blastomere biopsy and multiplex nested PCR analysis with simultaneous testing for p53 tumor-suppressor gene mutation and linked polymorphic markers, allowing preselecting and transferring back to the patient only mutation-free embryos (Fig. 13). A singleton pregnancy and birth of a mutation-free child resulted, and the child is currently healthy and free from the mutation predisposing to LFS.
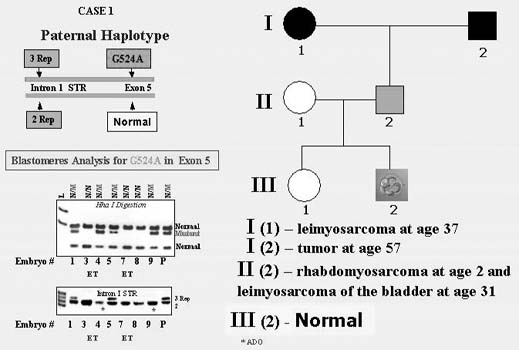{kind=link}
At present, PGD is also applied for other cancers, including familial adenomatous polyposis coli, Von Hippel Lindau syndrome, retinoblastoma, neurofibromatosis types I and II, and familial posterior fossa brain tumor.31 A few dozens of PGD cycles were performed, resulting in preselection and transfer of mutation- free embryos in the majority of them, which yielded unaffected clinical pregnancies and birth of healthy children. Despite the controversy of PGD use for late-onset disorders, the data demonstrate the usefulness of this approach as the only acceptable option for at-risk couples to avoid the birth of children with inherited predisposition to cancer and to have a healthy child (for the updated experience see25).
Other Late-Onset Disorders With Genetic Predisposition
One of the first experiences of PGD for late-onset disorders was PGD for genetic predisposition to one of the forms of Alzheimer disease (AD)32 caused by an autosomal-dominant familial predisposition to a presenile form of dementia, determined by nearly completely penetrant autosomal-dominant mutation in the amyloid precursor protein (APP) gene, for which no treatment is available despite a possible predictive diagnosis. A 30-year-old women had no signs of AD but was a carrier of V717L mutation, resulting from G-to-C substitution in exon 17 of the APP gene. The predictive testing in the patient was performed because of the early-onset AD in her sister carrying this mutation in whom symptoms of AD developed at age 38. This sister is still alive, but her cognitive problems progressed to the point that she was placed in an assisted living facility. Her father had died at age 42 and also had a history of psychological difficulties and marked memory problems. V717L mutation was also detected in one of her brothers who experienced mild short-term memory problems as early as age 35, with a moderate decrease in memory, new learning, and sequential tracking over the next 2–3 years. The other family members, including one brother and two sisters, were asymptomatic, although predictive testing was performed only in sisters, who appeared to be free from mutation in APP gene (Fig. 14).
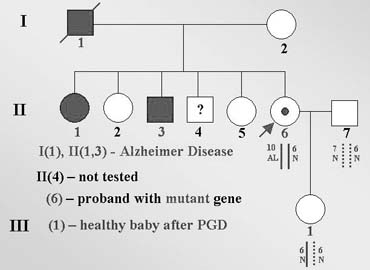{kind=link}
PGD was performed by DNA analysis of PB1 and PB2 to preselect and transfer back to the patient only the embryos resulting from mutation-free oocytes. Based on both mutation and the linked marker analysis, unaffected embryos resulting from mutation-free oocytes were preselected for transfer back to the patients, resulting in a singleton clinical pregnancy and birth of an unaffected mutation-free child.
PGD, therefore, provides a nontraditional option for patients who may wish to avoid the transmission of the mutant gene predisposing to the late-onset disorders in their potential children. Because such diseases present beyond early childhood or later and may not be expressed in 100% of the cases, the application of PGD for this group of disorders is still controversial. However, for diseases with no current prospect for treatment arising despite presymptomatic diagnosis and follow-up, PGD may be offered as the only relief for the at-risk couples (for the updated experience see 25).
The first PGD for maternal–fetal incompatibility resulting in the healthy pregnancy outcome was performed for Kell (K1) genotype, which is one of the major antigenic systems in human red blood cells, comparable in importance with RhD, causing maternofetal incompatibility leading to severe hemolytic disease of the newborn (HDN) in sensitized mothers.33 K1 allele is present in 9% of the populations, in contrast to its highly prevalent allelic variant K2. The gene is located on chromosome 7 (7q33), consisting of 19 exons with the only C-to-T base substitution in exon 6 in K1 compared with K2 antigen.
In cases of pregnancy by the K1 fetus in the K2 mother, antibodies to K1 may be developed, leading to maternofetal incompatibility causing severe HDN. Although prenatal diagnosis is available for identification of pregnancies at risk for HDN, this may not always prevent the potential complications for the fetus, such as stillbirth or neonatal death, making PGD a possible option for preventing both Kell and Rhesus hemolytic diseases.
PGD for Kell disease was performed for two at-risk couples with a history of neonatal death in previous pregnancies caused by HDN. The preselection and transfer of the embryos free from K1 allele of KEL gene was possible in each case, yielding a clinical pregnancy and the birth of healthy twins, confirmed to be free of K1 allele (Fig. 15).
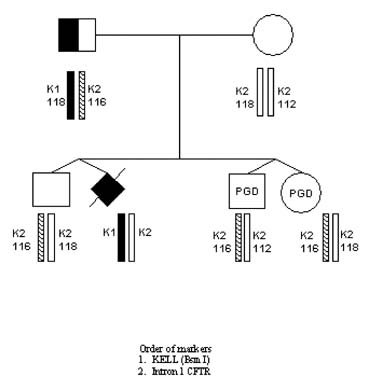{kind=link}
A number of attempts have also been undertaken to perform PGD for Rhesus disease, which, however, has not yet resulted in a clinical pregnancy.34 Both of these conditions are quite prevalent, taking into consideration the approximately 15% frequency for RhD and 9% for KEL antigen, presenting the risk for alloimmunization that may lead to HDN in some of the at-risk couples. Therefore, PGD may be a useful option for these couples to avoid the establishment of the RhD or K1 pregnancy in the sensitized mothers.
Although the at-risk pregnancies detected by prenatal diagnosis may be treated by an intrauterine transfusion, the potential complication for the fetus cannot be completely excluded even after this procedure. Pregnancy termination in such cases will also be unacceptable, because the antibodies to K1, for example, are developed only in 5% of persons obtaining incompatible blood. However, some of the at-risk couples have had such unfortunate experience with HDN, resulting in neonatal death as in both of our couples, that they regard PGD as their only option to plan another pregnancy. This makes PGD attractive for patients at risk for alloimmunization, although such conditions have rarely been an indication for prenatal diagnosis.
Congenital malformations are highly prevalent (29.3/1000 live births) and are usually sporadic. However, with progress of the human genome project, an increasing number of inherited forms are being described, which therefore may be avoided through PGD. For example, sonic hedgehog (SHH) gene mutation, for which the first PGD has recently been performed,22 causes the failure of cerebral hemispheres to separate into distinct left and right halves and leads to holoprosencephaly (HPE), which is one of the most common developmental anomalies of forebrain and midface. Although most HPE are sporadic, familial cases are not rare, with clear autosomal-dominant inheritance.
A great intrafamilial clinical variability of HPE from alobar HPE and cyclopia, to cleft lip and palate, microcephaly, ocular hypertelorism, and even normal phenotype, suggests the interaction of SHH gene with other genes expressed during craniofacial development and the possible involvement of environmental factors. This may explain the fact that almost one third of carriers of SHH mutations may be clinically unaffected. Therefore, even in familial cases, the detection of SHH mutations in prenatal diagnosis might not justify pregnancy termination, making PGD a more attractive option for couples at risk for producing a progeny with HPE, as demonstrated in the first PGD for this mutation.22
This couple presented for PGD because of the previous two children with the clinical signs of HPE. One of them, a female with severe HPE and cleft lip and palate, died soon after birth. Both the child and the parents were chromosomally normal, but DNA analysis in the child's autopsy material demonstrated the presence of SHH nonsense mutation caused by GAG-to-TAG sequence change, leading to premature termination of the protein at position 256 (Glu256 → stop). The same mutation was found in their 5-year-old son who was born after a full-term normal pregnancy. This child has less severe facial dysmorphisms, which included microcephaly, Rathke's pouch cyst, single central incisor, and choanal stenosis. There was also clinodactyly of the fifth fingers and curved-in fourth toes bilaterally. The child's growth was slow in the first 2 years, but thereafter he has been maintaining a reasonably good growth (Fig. 16).
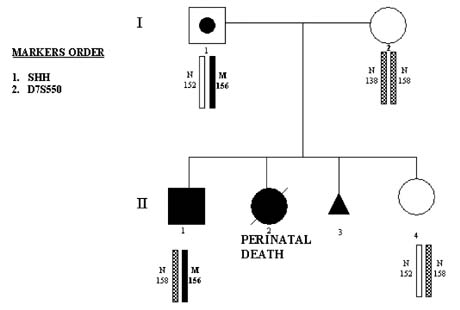{kind=link}
PGD was performed by blastomere biopsy and multiplex nested PCR analysis involving specific mutation testing simultaneously with linked marker analysis (Table 3). Of nine tested embryos, four embryos were free of mutant gene, from which two were transferred back to patient, resulting in a singleton pregnancy and birth of a healthy child after confirmation of the mutation-free status by amniocentesis (see Fig. 16). Similar approach was used for PGD of Crouson syndrome.35
The data suggest the clinical usefulness of PGD for familial cases of congenital malformations. Because of high prevalence of congenital anomalies, the approach may have practical implications for the at-risk couples as a preventive measure to be used in genetic practices (for the updated experience see25).
Preimplantation HLA Matching Combined With PGD
Preimplantation HLA-matching was first introduced in combination with mutation analysis for Fanconi anemia with the objective of establishing an unaffected pregnancy yielding a potential donor progeny for transplantation in an affected sibling.36 This resulted in a clinical pregnancy and birth of an unaffected child whose cord blood was transplanted to the affected sibling, thus saving her life (Fig. 17).
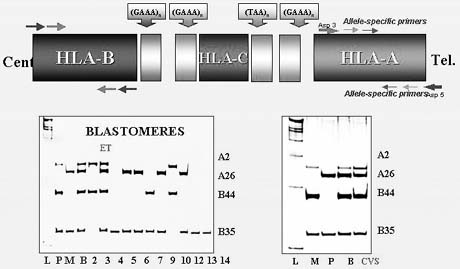{kind=link}
The strategy would not likely be clinically acceptable through traditional prenatal genetic diagnosis because of a possible clinical pregnancy termination after HLA-matching. However, PGD for such purpose should be acceptable, because only a limited number of the embryos are usually preselected for transfer, which in this case will represent unaffected embryos with a perfect match for affected siblings in need of transplantation. Because the multiplex single cell PCR used in PGD presents the opportunity for combined PGD and HLA testing, it has become a useful way to preselect an embryo that may be an HLA-match to the affected sibling requiring stem cell transplantation.
The method has currently been applied for the HLA-genotyping in two dozen cycles in combination with PGD for thalassemia, Fanconi anemia, hyper immunoglobulin M syndrome, X-linked adrenoleukodystrophy, and Wiskott-Aldrich syndrome, confirming the usefulness of preimplantation HLA-matching as part of PGD, with prospect of application of this approach to other inherited conditions also requiring an HLA-compatible donor for bone marrow transplantation.18, 25, 37, 38, 39, 40, 41, 42 This provides a realistic option for the couples desiring to avoid the birth of an affected child, together with the establishment of a healthy pregnancy, potentially providing an HLA-matched progeny for treatment of an affected sibling.
CONCLUSION
Important ethical issues have been raised with an increasing use of PGD for late onset disorders with genetic predisposion and preimplantation HLA typing to produce an HLA compatible donor for treating a family member with fatal bone marrow disease or cancer, requiring a stem cell transplantation.43, 44, 45, 46, 47 Although there is no actual difference in the application of PGD for the latter conditions, the controversy can be explained by the fact that in traditional prenatal diagnosis if the fetus would be found to carry the gene predisposing to late-onset disorder or to be HLA unmatched, the couple would have to make an extremely difficult decision of pregnancy termination, which could hardly be justified by such findings. Alternatively, PGD technology allows genetic testing of human eggs and embryos before pregnancy is established, making totally realistic to establish only HLA matched or potentially normal pregnancies without genetic predisposition to late onset common disorders.
In any case, as seen from this review, PGD is now becoming an established clinical option in reproductive medicine and is applied using separate consent forms and the research protocols approved by Institutional Ethics Committees. The number of apparently healthy children born after PGD is now a few thousand, showing that there is no evidence of any incurred adverse effect. However, these protocols would still require confirmatory CVS or amniocentesis and a follow-up monitoring of its safety and accuracy. Although PGD will help solve some of the long-standing ethical problems, such as the abortion issue (which will be avoided as a result of this new approach), others could become a serious obstacle, particularly those related to designer babies. These considerations are highly relevant to the subject of PGD, as well as to any other new methods as we proceed with further development of appropriate technology for controlling genetic disability.
REFERENCES
Verlinsky Y, Kuliev A: Atlas of Preimplantation Genetic Diagnosis. London, NY, Parthenon Publishing Group, 2000 |
|
Verlinsky Y, Kuliev A. Atlas of Preimplantation Genetic Diagnosis. Taylor and Francis, London, N.Y, 2005 |
|
Verlinsky Y, Cohen J, Munne S, Gianaroli L, Simpson JL, Ferraretti AP, Kuliev A. Abstract. Over a decade of experience with preimplantation genetic diagnosis: a multicenter report. Fertil Steril. 2004 Aug;82(2):292-4. |
|
Kuliev A, Verlinsky Y. 2008 Technological developments in preimplantation genetic diagnosis to improve the accuracy and range. Reproductive BioMedicine Online 5, 296-301. |
|
International Working Group on Preimplantation Genetics (IWGPG): Preimplantation Genetic Diagnosis--Experience of Three Thousand Clinical Cycles. Report of the 11th Annual Meeting International Working Group on Preimplantation Genetics, in conjunction with 10th International Congress of Human Genetics, Vienna, May 15, 2001 Reprod BioMed Online 3:49-53, 2001 |
|
Kuliev A, Verlinsky Y: Current features of preimplantation genetic diagnosis. Reprod BioMed Online 5:296-301, 2002 |
|
ESHRE Preimplantation Genetic Diagnosis Consortium: Data Collection III, May 2002. Hum Reprod 17:233-246, 2002 |
|
Preimplantation Genetic Diagnosis International Society (PGDIS) 2008 Guidelines for good practice in PGD: program requirements and laboratory quality assurance. Reproductive BioMedicine Online 16, 134-147 |
|
Rechitsky S, Verlinsky O, Amet T et al: Reliability of preimplantation diagnosis for single gene disorders. Mol Cell Endocrinol 183:S65-S68, 2001 |
|
Rechitsky S, Strom C, Verlinsky O et al: Allele drop out in polar bodies and blastomeres. J Assist Reprod Genet 15:253-257, 1998 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Polar body based preimplantation diagnosis for X-linked genetic disorders. Reprod BioMed Online 4:38-42, 2002 |
|
Kuliev A, Rechitsky S, Verlinsky O et al: Birth of healthy children after reimplantation diagnosis of thalassemias. J Assist Reprod Gent 16:207-211, 1999 |
|
Rechitsky S, Verlinsky O, Kuliev A et al: Preimplantation genetic diagnosis for familial dysautonomia. Reprod BioMed Online 6:488-496, 2003 |
|
Sermon K, Seneca S, De Rycke M et al: PGD in the lab for triplet diseases-myotonic dystrophy, Huntington's disease and Fragile-X syndrome. Mol Cell Endocrinol 183:S77-S85, 2001 |
|
Katz MG, Mansfield J, Gras L et al: Diagnosis of trisomy 21 in preimplantation embryos by single-cell DNA fingerprinting. Reprod BioMed Online 4:43-50, 2002 |
|
Harper J, Wells D, Piyamongkol W et al: Preimplantation genetic diagnosis for single gene disorders: experience with five single gene disorders. Prenat Diagn 22:525-533, 2002 |
|
Ray PF, Frydman N, Attie T et al: Birth of healthy twins after preimplantation diagnosis of Cystic fibrosis combined with gender determination. Mol Hum Reprod 8:688-694, 2002 |
|
Verlinsky Y, Rechitsky S, Verlinsky O, Ozen S, Beck R, Kuliev A. 2004a Preimplantation genetic diagnosis for polycystic kidney disease PKD Fertil Steril 82:926-929 |
|
Alterescu G, Brooks B, Margalioth EJ, Eldar-Geva T, Levy-Lahad E, Renbaum P 2007 Simultaneous preimplantation genetics diagnosis for Tay Sachs and Gaucher disease. Reproductive BioMedicine Online 15, 83-88 |
|
Treff NR, Su J, Mavrianos J, Bergh PA, Miller KA, Scot RT. 2007 Accurate 23 chromosome aneuploidy screening in human blastomeres using single nucleotide polymorphism (SNP) microarrays. Fertil Steril 88 (Suppl 1), S1 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for long-chain 3-hydroxyacyl-coa dehydrogenase deficiency. Reprod BioMed Online 2:17-19, 2001 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation Diagnosis for Sonic Hedgehog Mutation Causing Familial Holoprosencephaly. N Engl J Med 348:1449-1454, 2003 |
|
International Working Group on Preimplantation Genetics (IWGPG): Tenth Anniversary of Preimplantation Genetic Diagnosis: Report of the 10th Annual Meeting International Working Group on Preimlantation Genetics, in association with 3rd International Symposium on Preimplantation Genetics, Bologna, Italy, June 23, 2000. J Assist Reprod Genet 18:66-72, 2001 |
|
Kuliev A, Rechitsky S, Verlinsky Y. 2007 Preimplantation Diagnosis. Nature Encyclopedia of Life Sciences. A002507 |
|
Verlinsky Y. and Kuliev A. 2006 Practical Preimplantation Genetic Diagnosis. Springer, London, N.Y |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for ornithine transcarbamilase deficiency. Reprod BioMed Online 1:45-47, 2000 |
|
Ray P, Harper J, Ao A et al: Successful preimplantation genetic diagnosis for sex linked Lesch-Nyhan syndrome using specific diagnosis. Prenat Diagn 19:1237-1241, 1999 |
|
Ray PF, Gigarel N, Bonnefont JP et al: First specific preimplantation genetic diagnosis for ornithine transcarbamylase deficiency. Prenat Diagn 20:1048-1054, 2000 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for PKU. Fertil Steril 76:346-349, 2001 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for p53 tumor suppressor gene mutations. Reprod BioMed Online 2:102-105, 2001 |
|
Rechitsky S, Verlinsky O, Chistokina A et al: Preimplantation genetic diagnosis for cancer predisposition. Reprod BioMed Online 5:148-155, 2002 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for early onset Alzheimer disease Caused by V717L Mutation. J Am Med Assoc 287:1018-1021, 2002 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for Kell genotype. Fertil Steril 80:1047-1051, 2003 |
|
Van Den Veyver IB: Prenatal and Preimplantation Genetic Diagnosis for RhD Alloimmunization, pp 181–185. 10th International Conference on Prenatal Diagnosis and Therapy. Barcelona (Spain), June 19–21, 2000. Barcelona, University of Barcelona, 2000 |
|
Abou-Sleiman PM, Apessos A, Harper JC et al: Pregnancy following preimplantation genetic diagnosis for Crouson syndrome. Mol Hum Reprod 8:304-309, 2002 |
|
Verlinsky Y, Rechitsky S, Verlinsky O et al: Preimplantation diagnosis for Fanconi anemia combined with HLA matching. JAMA 285:3130-3133, 2001 |
|
Rechitsky S, Kuliev A, Tur-Kaspa I, Morris R, Verlinsky Y. 2004 Preimplantation HLA typing with preimplantation genetic diagnosis. Reproductive BioMedicine Online 6:488-493. |
|
Kahraman S, Karlilaya G, Sertyel S, Karadayi H, Findicli N, Oncu N. 2004 Clinical Aspects of Preimplantation Genetic Diagnosis of Single Gene Disorders Combined with HLA Typing. Reproductive BioMedicine Online 9: 529-532. |
|
Van deVelde H, Georgiou I, De Rycke M et al. 2004 Novel universal approach for preimplantation genetic diagnosis of β-thalassemia in combination with HLA matching of embryos. Hum Reprod 19:700-708 |
|
Kuliev A, Rechitsky S, Verlinsky O, et al. 2005. Preimplantation diagnosis and HLA typing for hemoglobin disorders. Reproductive BioMedicine Online 11: 362-370 |
|
Rechitsky S, Kuliev A, Sharapova T, et al. 2006 Preimplantation HLA typing with aneuploidy testing. Reprod BioMedicine Online 12: 81-92. |
|
Verlinsky Y, Rechitsky S, Sharapova T et al. 2006 Preimplantation diagnosis for immnudeficiencies. Reprod BioMed Online 4, 38-42 |
|
Damewood MD: Ethical implications of a new application of preimplantation diagnosis. JAMA 285:3143-3144, 2001 |
|
Towner D, Loewy RS: Ethics of preimplantation diagnosis for a woman destined to develop early-onset Alzheimer disease. JAMA 287:1038-1040, 2002 |
|
Edwards RG: Social and ethical issues of PGD, cloning and gene therapy. Reprod BioMed Online 6:170-180, 2003 |
|
Robertson JA: Extending preimplantation genetic diagnosis: the ethical debate. Hum Reprod 18:465-471, 2003 |
|
Edwards RG. Ethics of PGD: thoughts on the consequences of typing HLA in embryos. Reproductive BioMedicine Online 2004; 9:222-224. |